Download

1 / 24
310 likes | 1.64k Views
OSTEOGENESIS IMPERFECTA AND ACHONDROPLASIA. JENNIFER ABARA 0612 MD5. CONTENTS. INTRODUCTION/OVERVIEW EPIDEMIOLOGY ETIOLOGY PATHOPHYSIOLOGY CLINICAL PRESENTATION DIAGNOSIS/WORKUP TREATMENT/MANAGEMENT COMPLICATIONS REFERENCES. OSTEOGENESIS IMPERFECTA. Hereditary bone disorder

E N D
OSTEOGENESIS IMPERFECTA AND ACHONDROPLASIA JENNIFER ABARA 0612 MD5
CONTENTS • INTRODUCTION/OVERVIEW • EPIDEMIOLOGY • ETIOLOGY • PATHOPHYSIOLOGY • CLINICAL PRESENTATION • DIAGNOSIS/WORKUP • TREATMENT/MANAGEMENT • COMPLICATIONS • REFERENCES
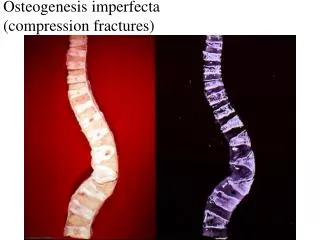
OSTEOGENESIS IMPERFECTA • Hereditary bone disorder • one of the most common skeletal dysplasias. • Also called Lobstein disease, brittle-bone disease, blue-sclera syndrome, and fragile-bone disease. • Group of hereditary disorders caused by defective synthesis of type I collagen • Autosomal dominant disorder
EPIDEMIOLOGY • The overall incidence of OI is approximately 1 case for every 20,000 live births; however, the mild form is underdiagnosed, and the actual prevalence may be higher. • OI can present at any age, although the age when symptoms (ie, fractures) begin varies widely. Patients with mild forms may not have fractures until adulthood, or they may present with fractures in infancy. • OI is equally common in males and females.
PATHOGENESIS • Molecular pathology underlying OI involves gene mutations in the coding sequences for α1 or α2 chains of type I collagen • Type 1 collagen is a triple helix formed by 2 copies of the alpha1 chain and 1 copy of the alpha2 chain. The COL1A gene on chromosome 17 encodes the pro-alpha1 chain, and the COL2A gene on chromosome 2 encodes the pro-alpha2 chain. • Because successful collagen synthesis and extracellular export require formation of a complete and intact triple helix, any primary defect in a collagen chain tends to disrupt the entire structure and results in its premature degradation -- Dominant negative mutation & disastrous phenotype
TYPES OF OI • 4major subtypes/clinical phenotypes, with an extremely broad range of clinical outcomes. The fundamental abnormality in all forms of OI is too little bone, resulting in extreme skeletal fragility.
CLINICAL FEATURES • Generalized osteopenia (Brittle bones)→ recurrent fractures & skeletal deformity of virtually every bone in the body. • Blue sclera— Abnormal thin sclera. Underlying choroidal veins becomes visible • Hearing loss (Conductive and sensorineural) - About 50% of patients with type I OI have deafness by age 40 years. • Dentinogenesisimperfecta • Abnormally thin dermis, leading to easily bruised skin • Limb deformities • Triangular facies
DIAGNOSIS/WORKUP • Collagen synthesis analysis - performed by culturing dermal fibroblasts obtained during skin biopsy. • Prenatal ultrasonography - capable of detecting limb-length abnormalities at 15-18 weeks’ gestation • Plain Radiography - Radiologic features commonly seen include fractures, Excessive callus formation and popcorn bones, Deformities of the thoracic cage,etc • Densitometry - Bone mineral density, as measured with DEXA, is low in children and adults with OI regardless of severity.
DIFFERENTIAL DIAGNOSIS • May be confused with Child Abuse; though it must be kept in mind that the 2 can also coexist. • Osteoporosis • Osteopetrosis • Scurvy • Rickets • Achondroplasia
TREATMENT/MANAGEMENT • Surgical correction of deformities - reserved for functional improvement. Remains a pillar of treatment for patients with OI. • Physiotherapy • Use of orthotic support and devicesto assist mobility (eg, wheelchairs) • Intramedullary Rod Placement. • Pharmacologic Therapy – Bisphosphonates (egIV pamidronateis commonly given in a dosage of 7.5 mg/kg/y at 4-6 month intervals), Teriparatide (recombinant human form of parathyroid hormone) • Currently, medical treatments aimed at increasing bone mass and strength are gaining popularity.
ACHONDROPLASIA • Heterogeneous group of disorders characterized by intrinsic abnormalities in the growth and/or remodeling of cartilage and bone. • AD; 80% new mutations(sporadic mutations); related to increased paternal age • Most common form of inherited dwarfism/ major cause of dwarfism • most common type of short-limb disproportionate dwarfism. • Affects the skull, spine, and extremities in varying degrees • The characteristic features of achondroplasia are apparent at birth.
epidemiology • Approximately 10,000 individuals are estimated to have achondroplasia in the US • Achondroplasia is the most common skeletal dysplasia. It affects about 1 in every 40,000 children. • Approximately 150,000 persons have achondroplasia worldwide. (Total=190,000) • Achondroplasia occurs in all the races with equal frequency. • Achondroplasia occurs with equal frequency in males and females.
PATHOGENESIS • The underlying etiology is an activating (point) mutation in the fibroblast growth factor receptor 3 (FGFR3) on chromosome 4 that results in its constitutive or inappropriate activation. • Unfortunately, activated FGFR3 inhibits chondrocyte proliferation; as a result, the normal epiphyseal growth plate expansion is suppressed and there’s decreased endochondral bone formation and premature ossification of the growth plates --- long bone growth is severely stunted. • Absence or attenuation of zone of proliferative cartilage
CLINICAL PRESENTATION • Long bones are short and thick → short extremities → Dwarfism • Head and torso are grossly normal/Cranial & vertebral bones are spared • Normal intelligence/Mental function, life span, and reproductive ability/fertility • Kyphoscoliosis → Respiratory problems like corpulmonale • Hip problems • Bowing of the legs due to joint laxity • Lordotic (Sway-backed) posture/ Excessive lumbar lordosis • Flared metaphyses, Shortened diaphysis
Diagnosis/work up • Cytogenetics- Plasma can be analyzed for the FGFR3 mutation in the mother when a short-limb skeletal dysplasia is diagnosed prenatally on ultrasound. • Imaging studies (CT scan, MRIs) - Primary radiographic criteria for diagnosis include Decrease in interpedicular distance in the lumbar spine, Short, broad neck of femur, Shortening of long tubular bones, with metaphysealflaring etc • Prenatal ultrasonography - Later in the pregnancy (from 20-24wks), ultrasonography can detect short-limb dysplasia
TREATMENT/MANAGEMENT • Medical Care – The availability of somatotropin (recombinant human growth hormone) has revolutionized the treatment of short stature. Growth hormone is currently being used to augment the height of patients with achondroplasia. • Surgical Care - Most of the orthopedic problems encountered in patients with achondroplasia are related to the spine. They include: Craniocervical stenosis, thoracolumbar kyphosis, spinal stenosis, angular deformities of the lower extremities, and lengthening of the short extremities.
References • http://emedicine.medscape.com/article/OsteogenesisImperfecta/1256726-overview • http://emedicine.medscape.com/article/Achondroplasia/1258401-overview • Robbins Basic Pathology, 8th Edition • Pathoma • Kaplan Pathology