Download
1 / 14
140 likes | 285 Views
Cystic Fibrosis (CF) is a genetic disorder characterized by thick, sticky mucus build-up in the lungs and digestive system, leading to severe respiratory infections and digestive issues. CF is caused by a defective gene, affecting mostly those of Northern European descent. Early signs include salty skin, persistent cough, and digestive difficulties. While there’s no prevention for CF, treatments like antibiotics, inhaled medications, and special diets improve quality of life, with life expectancy significantly increased since the 1950s.

E N D
Anthony Bui & Nhan Nguyen Jacobs Anatomy – 4 12 April 2011 Respiratory Diseases
What is Cystic Fibrosis? • Cystic Fibrosis is an inherited disease that causes the build-up of thick, sticky mucus in the lungs and digestive track. • The collection of the mucus results in life-threatening lung infections and serious digestion problems. • The disease may also affect the sweat glands and a man’s reproductive system. =[
What causes it? • Cystic Fibrosis is caused by a defective gene which causes the body to produce abnormally thick and sticky fluid, called mucus. • The mucus builds up in the breathing passages of the lungs and in the pancreas. • There is little known about the CFTR protein that causes CF, because it is hydrophobic and difficult to crystallize.
Who will have it? • Millions of Americans carry the Cystic Fibrosis gene, but do not have any symptoms (they are simply carriers). • The disease is more common among those of Northern or Central European descent. • Most children with CF are diagnosed by age two, while a rare few are not diagnosed until age 18 or older.
What are some early signs? • Very salty-tasting skin; • Persistent coughing, at times with phlegm; • Wheezing or shortness of breath; • Frequent greasy, bulky stools or difficulty in bowel movements ; and • Ongoing diarrhea.
A few later signs… • Frequent lung infections; • Poor growth/weight gain in spite of a good appetite; (chronic malnutrition) • Inability to digest and absorb vital nutrients; (because of buildup in pancreas) and • Death, which is the most severe effect of Cystic Fibrosis.
Prognosis. • In the 1950s, few children with cystic fibrosis lived to attend elementary school. • Today, most children are fairly healthy until they reach adulthood. • They are able to participate in most activities and find employment easily. • BUT, the lung disease eventually worsens to the point where the person is disabled. Now, the average life span is 35 years, which is a dramatic increase since the 1950s.
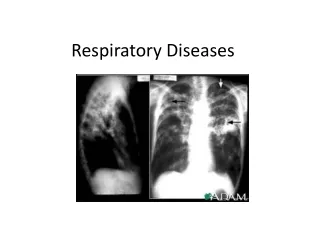
How to diagnose it • The ImmunoreactiveTrypsinogen (IRT) test is a standard screening test for CF, a high level of IRT suggests possible CF and requires further testing. • Sweat chloride test is the standard diagnostic test for CF. • Chest x-ray or CT Scans • Fecal fat tests • Lung Function tests • Measurement of pancreatic function • Secretin Stimulation tests
http://www.dimensionsguide.com/wp-content/uploads/2010/01/X-ray-Machine.jpghttp://www.dimensionsguide.com/wp-content/uploads/2010/01/X-ray-Machine.jpg
How to treat Cystic Fibrosis Treatments include: • Antibiotics to prevent and treat infections • Inhaled medicines to help open airways • Special diet high in proteins and calories • Pancreatic enzymes to help absorb nutrients • Vitamin Supplements!
http://newcareerasacoach.com/wp-content/uploads/2011/03/cysticfibrosis_395.jpghttp://newcareerasacoach.com/wp-content/uploads/2011/03/cysticfibrosis_395.jpg
Preventative measures • Unfortunately, there is no way to prevent Cystic Fibrosis. • Cystic Fibrosis is totally hereditary. • The Cystic Fibrosis gene affects 1 in 29 Caucasian Americans, mostly of Northern European descent.
Works Cited! Content Sources • http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001167/ • http://www.cff.org/AboutCF/ • http://www.ehow.com/facts_5006996_what-effects-cystic-fibrosis.html • http://cystic-fibrosis.emedtv.com/cystic-fibrosis/early-symptoms-of-cystic-fibrosis.html Image Sources • http://www.eradimaging.com/images/cystic_fibrosis_2008/cystic_fibrosis_fig1.jpg • http://www.wagerrun.com/wp-content/uploads/2010/06/CF-Logo.jpg • http://newcareerasacoach.com/wp-content/uploads/2011/03/cysticfibrosis_395.jpg • http://parenting.leehansen.com/downloads/clipart/halloween/images/skeleton.jpg • http://www.kdheks.gov/safekids/images/kidsHome.jpg • https://www.achooallergy.com/blog/images/mucus-rules.jpg