Download

1 / 31
310 likes | 469 Views
Functional Gene Clustering via Gene Annotation Sentences, MeSH and GO Keywords from Biomedical Literature. Dr. N. JEYAKUMAR, M.Sc., Ph.D., Bioinformatics Centre School of Biotechnology Madurai Kamaraj University Madurai – 625021, INDIA. Purpose & Goals.

E N D
Functional Gene Clustering via Gene Annotation Sentences, MeSH and GO Keywords from Biomedical Literature Dr. N. JEYAKUMAR, M.Sc., Ph.D., Bioinformatics Centre School of Biotechnology Madurai Kamaraj University Madurai – 625021, INDIA
Purpose & Goals • Extracting gene specific functional ‘keywords’ from biological literature • From full-abstracts • Gene specific sentences • Augment extracted keywords with MeSH and GO keywords related to gene • Compare the accuracy of results with a test data set in various keyword extraction methods • Full-abstracts • Gene specific sentences • Gene specific sentences + MeSH keywords • Gene specific sentences+ MeSH and GO keywords • Use the keyword extraction method to cluster the differentially expressed gene clusters in a microarray experiments
Outline ? • Part I: Text mining and keyword extraction from literature • Our text mining methodology • Part II: Applications to microarrays • Functional keyword clustering of microarray data • Two Parts: I, and II
Text Mining:Introduction and overview • Text mining aims to identify non-trivial, implicit, previously unknown, and potentially useful patterns in text (e.g. classification system, association rules, hyphothesis etc.) • includes more established research areas such as • information retrieval (IR), • natural language processing (NLP), • information extraction (IE), • and traditional data mining (DM) • relevant to bioinformatics because of • explosive growth of biomedical literature (e.g. MEDLINE – 15 million records) • availability of some information in textual form only, e.g. clinical records
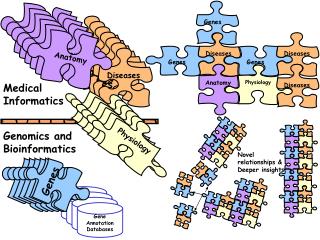
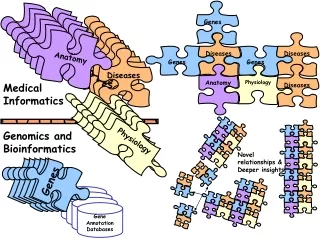
Text Mining:System Architecture Experimental design of gene clustering with sentences-level, MeSH and GO keywords
Text Mining:Keyword Extraction from Biomedical Literature Steps to extract sentence-level keywords • Gene - Synonym dictionary – A special gene name synonym name dictionary was created for human genes using Entrez-Gene • Gene-name normalization - This process replaces all the gene names in the abstract with its unique canonical identifier (Entrez gene ID) using the gene-synonym dictionary specially constructed for this study. • Sentence filtering – using corpus specific the regular expression as the following example ($gene @{0,6} $action (of|with) @{0,2} $gene) • extracts sentences that match the structure shown below the expression. The notational construct ‘AB ...’ is interpreted as ‘A followed by B followed by ...’. • gene name 0-6 wordsaction verb ‘of’ or ‘with’ 0-2 wordsgene name • Keyword extraction. – Next slide
Text Mining:Keyword Extraction from biomedical literature Table 1. An example set of regular expressions as nouns describing agents and agents, and passive and active verbs
Text Mining:Keyword Extraction from Biomedical Literature Keyword extraction Example • Sentence: • BRCA1 physically associates with p53 and stimulates its transcriptional activity. • Brill-POS-tagged sentence: • BRCA1/NNP physically/RB associates/VBZ with/IN p53/NN and/CC stimulates/VBZ its/PRP$ transcriptional/JJ activity/NN ./. • Sentence keywords: • associates, stimulates, transcription activity • Sentence keywords after manual curation: • transcription activity
Text Mining:MeSH Keyword Extraction • MeSH keywords • MeSH keywords are subject index terms assigned to each scientific literature by the Natural Library of Medicine (NLM) for purpose of subject indexing and searching the journal articles via PubMed. • MeSH keyword extraction • Extracted directly from gene specific abstracts via Perl scripts • MeSH keyword curation • Using a MeSH keywords stop words dictionary (e.g., human, DNA, animal, Support U.S Govt etc.). • For example the MeSH keywords associated with a gene ‘FOS’ in our gene list are ‘oncogene, felypressin, transcription-factor, thermo-receptors, DNA-binding, antibiosis, inflammatory-response, zinc-fingers, gene-regulation, and neuronal-plasticity’.
Text Mining:GO Keyword Extraction • GO keywords • Gene Ontology (GO) is a hierarchical organization of gene and gene product terms from various databases in which concepts at higher levels in the hierarchy are more general than those further down • GO keyword extraction • Out of the three GO annotation categories we included only molecular function and biological process and left out cellular component as it is less important for characterizing genes functions • Further, due to hierarchical nature of GO and multiple inheritance in the GO structure, we consider with every ancestor up to the level 2 in the GO tree • For example the GO keywords associated with the gene ‘FOS’ in our gene list are ‘protein-dimerization, DNA binding, RNA polymerase, transcription factor, DNA methylation, and inflammatory-response’.
Text Mining:Keyword Representation and Calculation of Numeric Vectors • This process is concerned with computing the numeric weight, wij, for each gene-keyword pair (gi, tj) (i = 1, 2, … n and j = 1, 2, … k) to represent the gene’s characteristics in terms of the associated keywords. • Common techniques for such numeric encoding include • Binary. The presence or absence of a keyword relative to a gene. • Term frequency. The frequency of occurrence of a keyword with a gene. • Term frequency / inverse document frequency (TF*IDF). The relative frequency of occurrence of a keyword with a gene compared to other genes
Text Mining:TF*IDF Weighting • Most weighting scheme in information retrieval and text classification method is the TFIDF (term frequency / inverse document frequency) weighting scheme. • TF(w,d) (Term Frequency) is the number of times word w occurs in a document d. • DF(w) (Document Frequency) is the number of documents in which the word w occurs at least once. • The inverse document frequency is calculated as • Where | D |is total number of documents in the corpus
Text Mining:Keyword Representation and Calculation of Numeric vectors • In our study, as the keywords are extracted from gene specific sentences but not from full abstracts, the number of keywords associated with each gene is small. • Further, the frequency of occurance of most keywords tended be one. • Therefore, the binary encoding scheme was adopted as illustrated in Table 2 . Table 2. Binary representation of gene * keywords
Text Mining:Gene Clustering • After, our binary coding scheme adopted in this study consists of numeric row vectors representing genes (via the associated biological functional keywords), and numeric column vectors representing annotation terms (via the associated genes) • Clustering can produce useful and specific information about the biological characteristics of sets of genes • Clustering: Partition unlabeled examples into disjoint subsets of clusters, such that: • Examples within a cluster are very similar • Examples in different clusters are very different • Discover new categories in an unsupervised manner.
Text Mining:Test Set and Evaluation • The test set contains 20 genes and 10 abstracts for each gene, resulting in a total of 200 abstracts in two cancer categories (Table 3) was used evaluate usefulness of our keyword extraction method Table 3. Test set of 20 human genes manually grouped in to two cancer categories
Text Mining:Evaluation • Full abstract keywords (baseline). Extracts gene annotation terms based on term frequencies * inverse document frequencies (TF*IDF) within the entire abstract without regard to sentence structure. • Sentence keywords. Extracts gene specific keywords based sentence-level processing. • Sentence + MeSH keywords. As in (2) above plus MeSH terms (see Section MeSH keywords extraction). • Sentence + MeSH + GO keywords. As in (2) above plus MeSH terms (see Section MeSH keywords extraction) and GO terms (see Section GO keyword extraction
Text Mining:Evaluation Results of various keyword extraction methods
Part II: Applications to Microarrays Functional keyword Clustering of genes resulting from microarray experiment
Applications to MicroarraysData and Analysis • As an illustrative example, our keyword extraction methods was applied to functional interpretation of cluster of genes that were found differentially expressed in a microarray experiment investigating the impact of two mitogenic protein Epidermal growth factor (EGF) and Sphingosine 1-phosphate (S1P) on glioblastoma cell lines • when compared to the resting state, 19 genes were significantly differentially expressed as a response to EGF, 35 genes as a response to S1P and 30 genes as a response to COM, i.e., combined stimuli of S1P and EGF. The three gene lists are referred to as G(EGF), G(S1P) and G(COM), respectively (Table 4).
Applications to MicroarraysData and Analysis Table 4. List of Differentially Expressed Genes
Applications to Microarrays Data and Analysis • Using these the three gene lists obtained from the microarray experiment (Table 6) as query in MEDLINE returned the three corresponding sets of abstracts A(EGF), A(S1P) and A(COM), respectively (Table 5). • The abstracts were processed with the keyword extraction method involving sentence-level augmented with MeSH and GO keywords • The resulting keywords were encoded in binary weighting scheme • The resulting representations were clustered using average linkage hierarchical clustering algorithm.
Applications to MicroarraysData and Analysis Table 5. Three sets of abstracts, A(EGF), A(S1P), and A(COM), retrieved via MEDLINE for this study
Applications to Microarrays Average Linkage Hierarchical Clustering Algorithm • Use average similarity across all pairs within the merged cluster to measure the similarity of two clusters. • Compromise between single and complete link. • Averaged across all ordered pairs in the merged cluster instead of unordered pairs between the two clusters.
Summary of analysis of EGF cluster Applications to Microarrays Results
Summary of analysis of S1P cluster Applications to Microarrays Results
Summary of analysis of COM cluster Applications to Microarrays Results
Conclusions • An important topic in microarray data mining is to bind transcriptionally modulated genes to functional pathways or how transcriptional modulation can be associated with specific biological events such as genetic disease phenotype, cell differentiation etc. • However, the amount of functional annotation available with each transcriptionaly modulated genes is still a limiting factor because not all genes are well annotated • Further, Jenssen et al. (2001) earlier compiled a network of human gene relationships from MEDLINE abstracts. These compiled relationships were then compared to the gene expression cluster results. This approach gives a very interesting result: functionally related genes can show totally different patterns, and hence belong to different clusters (Jenssen, et al.: A literature network of human genes for high-throughput analysis of gene expression, Nat.Genet., 28, 21-28, 2001)
Conclusions • Our gene functional keyword clustering/ grouping will enable to select functionally informative genes from differentially expressed genes for further investigations. • Our evaluation suggests that this approach will provide more specific and useful information than typical approaches using abstract-level information. This is particularly the case when the sentence-level terms are augmented by MeSH and GO keywords • As the current text mining scenario is on full-text mining As full-text contains large number of irreverent sentences compare to abstracts this approach is more appropriate for full-text study as it filters irrelevant sentences before clustering.
Eric G. Bremer, Brain Tumor Research Program, Children’s Memorial Research Center, Chicago, IL, USA, and James R. van Brocklyn, Division of Neuropathology, Department of Pathology, The Ohio State University, Columbus, Ohio, USA for the microarray data set Dr. Daniel Berrar, Bioinformatics Research Group, University of Ulster, UK Members of Bioinformatics Centre, Madurai Kamaraj University, India Dept of Biotechnology, Govt. of India for Bioinformatics facilities Acknowledgments