Download

1 / 19
190 likes | 411 Views
The Extraction of Single Nucleotide Polymorphisms and the Use of Current Sequencing Tools. Stephen Tetreault Department of Mathematics and Computer Science Rhode Island College Providence, RI. Single Nucleotide Polymorphisms.

E N D
The Extraction of Single Nucleotide Polymorphisms and the Use of Current Sequencing Tools Stephen Tetreault Department of Mathematics and Computer Science Rhode Island College Providence, RI
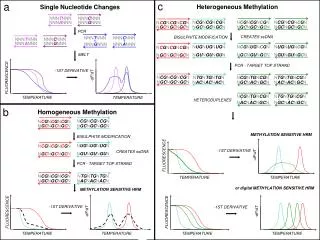
Single Nucleotide Polymorphisms • DNA sequence variation when a single nucleotide in the genome differs • SNPs are the majority of genetic variation • 1.4 million SNPs in a human genome • Two haploid genomes differing at 1 SNP per 1,331 bp • SNPs are crucial in the effort to personalize medicine
1000 Genomes Project • International consortium to create most complete catalog of human genetic variation • Sequencing is done using utilizing next generation sequencing technology (e.g. Solexa, 454, SOLiD) which is faster and less expensive • 3 steps of the project: • Detailed scanning of six participants • Less detailed scan of 180 participants • Partial scans of 1000 participants
1000 Genomes Project • 1000 Genomes Project Goals: • Discover genetic variants (SNPs, copy-number variants, indels) • Identify frequencies of the variant alleles and identify their haplotype backgrounds
Project Focus • Learning about the current state of sequencing tools • Learning how to use these tools and understanding the raw data • Creating a program to to extract the SNPs from the raw data and to calculate simple variant frequencies. • More advanced data analysis - to be discussed in future works section
Data and Tools • 1000 Genomes Project • ftp://ftp-trace.ncbi.nih.gov/1000genomes/ • MAQ 0.7.1 • http://sourceforge.net/projects/maq/files/ • SAMtools 0.1.5 • http://sourceforge.net/projects/samtools/files/
Sequencing • MAQ maps short reads to references and calls genotypes from the alignment • MAQ maps a read to the position where the sum of quality values of mismatched nucleotides is minimum • Issues with MAQ: • Very long run-time • Limited computing power slowed the program down
Sequencing • SAMtools was the alternative sequencing program. • It proved faster because it could utilize BAM (Binary SAM) files which are prealigned partial scans of the participant data. • MAQ had to align FASTA and FASTQ files, then change the MAP file into a Consensus file for SNP calling. • SAMtools allowed for SNP calling as MAQ did • SAMtools pileup function describes base pair information at each chromosomal position.
Sequencing • SAMtools pileup function describes base pair information at each chromosomal position.
Project Data • The raw data received through SAMtools pileup and consensus calling contains the following: chromosome, position, reference base, consensus base, consensus quality score, SNP quality score, maximum mapping quality score, number of reads mapped, read bases, and base qualities.
Phred Quality Scores • The consensus quality score and the SNP quality are Phred quality scores. • High accuracy of Phred scores helps ensure reliable SNP calling
Finding Higher Quality SNPs • Look at the number of reads covering the position with th SNP and discard those covered by three or fewer reads. • Consensus quality is important, but SNP quality is more important. Discard a SNP with a quality score lower than 20.
A Program for Extracting SNPs • Read in raw data line by line • Check for SNP of high quality • Differing reference and consensus base • SNP with a quality score of 20 or higher • Insert SNP as on object into array list (also stored in order of position) • Keep counts for variant frequency & update when SNP is found • Keep count of number of SNPs per 100,000 bases throughout chromosome 1
Results • Comparing variant frequencies: • Base change of A to G and of T to C were shown to be the most frequently occuring variations • Base change of C to G was least frequently occuring
Results • The number of SNPs occuring per 100,000 bases throughout chromosome 1 for participant NA07048
Results • The number of SNPs occuring per 100,000 bases for chromosome 1 of participant NA12273. The SNPs appear more clustered together in frequency when compared to NA07048.
Conclusion • Initial complications in data access and slow progress with MAQ were overcome. • SAMtools proved to be faster thus more efficient at sequencing and SNP calling when utilizing the prealigned partial BAM files
Future Work • FastPHASE is a program used for estimating missing genotypes and for reconstruction of haplotypes. • Implement advanced data analysis into program by calling genotypes from the reads and running fastPHASE to obtain corresponding haplotypes. • Look at chromosome 1 for an individual and look at the reads mapped covering that position and see what the bases are for that position to determine if the SNP is heterozygous or homozygous
Acknowledgment • Thank you to the Professor Yufeng Wu, Jin Zhang, the Computer Science and Engineering Department at University of Connecticut, and the National Science Foundation for making this project and the Bio-Grid REU possible.