Download

1 / 29
300 likes | 510 Views
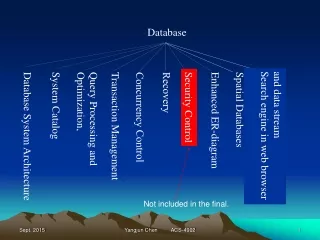
Glycan database. Database of molecules. Two models (of vocabularies) Proteins / Nucleic Acids Residues (+ modifications) Genbank / Swissprot Compounds Atoms & covalent bonds (SMILE/SMARTS language) Pubchem / ACS Glycans Residues: monosaccahrides (+ many modifications)

E N D
Database of molecules • Two models (of vocabularies) • Proteins / Nucleic Acids • Residues (+ modifications) • Genbank / Swissprot • Compounds • Atoms & covalent bonds (SMILE/SMARTS language) • Pubchem / ACS • Glycans • Residues: monosaccahrides (+ many modifications) • Branching nonlinear structure
Simplified molecular input line entry specification (SMILE) • Glucose • OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)1
Representation of glycans • Vocabulary • monosaccharides rather than atoms • Two challenges • Controlled vocabulary of monosaccharides • GlycoCT • From residues to molecules: glycan exchange format • GLYDE-II
Searching the glycan database: comparison • Glycan representation • tree vs. sequences • Glycan matching • exact vs. non-exact • Graph theoretic algorithm • alignment? Mutations are natural events. • Multiple glycan matching • Glycan pattern searching • Significance estimation
GLYDE standard • An XML based representation format for glycan structures • Inter-convertible with existing data represented using IUPAC or LINUCS. • GLYDE II: Incorporation of Probability based representation • Visualization: structures using GLYDE (XML) files GLYDE - An expressive XML standard for the representation of glycan structure. Carbohydrate Research, 340 (18), Dec 30, 2005.
Collaborative GlycoInformatics • Enable querying and export of query results in GLYDE format • Using GLYDE representation for disambiguation, mapping and matching GLYDE MonosaccharideDB <glyde> <residue> . . </residue> </glyde> SweetDB QUERY <glyde> <residue> . . </residue> </glyde> KEGG RESULT
Semantic GlcyoInformatics - Ontologies • GlycO: A domain ontology for glycan structures, glycan functions and enzymes (embodying knowledge of the structure and metabolisms of glycans) • Contains 600+ classes and 100+ properties – describe structural features of glycans; unique population strategy • URL: http://lsdis.cs.uga.edu/projects/glycomics/glyco • ProPreO: a comprehensive process Ontology modeling experimental proteomics • Contains 330 classes, 6 million+ instances • Models three phases of experimental proteomicsURL: http://lsdis.cs.uga.edu/projects/glycomics/propreo
GlycO taxonomy The first levels of the GlycO taxonomy Most relationships and attributes in GlycO GlycO exploits the expressiveness of OWL-DL. Cardinality constraints, value constraints, Existential and Universal restrictions on Range and Domain of properties allow the classification of unknown entities as well as the deduction of implicit relationships.
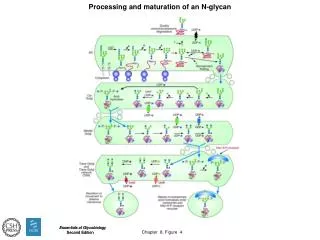
<Glycan> <aglycon name="Asn"/> <residue link="4" anomeric_carbon="1" anomer="b" chirality="D" monosaccharide="GlcNAc"> <residue link="4" anomeric_carbon="1" anomer="b" chirality="D" monosaccharide="GlcNAc"> <residue link="4" anomeric_carbon="1" anomer="b" chirality="D" monosaccharide="Man" > <residue link="3" anomeric_carbon="1" anomer="a" chirality="D" monosaccharide="Man" > <residue link="2" anomeric_carbon="1" anomer="b" chirality="D" monosaccharide="GlcNAc" > </residue> <residue link="4" anomeric_carbon="1" anomer="b" chirality="D" monosaccharide="GlcNAc" > </residue> </residue> <residue link="6" anomeric_carbon="1" anomer="a" chirality="D" monosaccharide="Man" > <residue link="2" anomeric_carbon="1" anomer="b" chirality="D" monosaccharide="GlcNAc"> </residue> </residue> </residue> </residue> </residue> </Glycan>
ProPreO • ProPreO: A process ontology to capture proteomics experimental lifecycle: • Separation • Mass spectrometry • Analysis • 330 classes • 110 properties • 6 million+ instances
Usage: Mass spectrometry analysis Manual annotation of mouse kidney spectrum by a human expert. For clarity, only 19 of the major peaks have been annotated. Goldberg, et al, Automatic annotation of matrix-assisted laser desorption/ionization N-glycan spectra, Proteomics 2005, 5, 865–875
Semantic Annotation of Experimental Data • Enables Ontology-mediated Disambiguation • Allows correlation between disparate entities using Semantic Relations P(S | M = 3461.57) = 0.6 P(T | M = 3461.57) = 0.4 Goldberg, et al, Automatic annotation of matrix-assisted laser desorption/ionization N-glycan spectra, Proteomics 2005, 5, 865–875
Graph Theoretic Basics • tree: an acyclic connected graph, whose vertices we refer to as nodes; • rooted tree: a tree having a specific node called the root, from which the rest of the tree extends. • children: nodes that extend from a node x by one edge are called the children of x; and conversely, x would be called the parent of these children; • Leaf: a node with no children; • Subtree: subtree of a tree T is a tree whose nodes and edges are subsets of those of T; • ordered tree: the rooted tree in which the children of each node are ordered; • labeled tree: a tree in which a label is attached to each node; • Forest: a set of trees • Oligosaccarides can be represented as labeled (monosaccahrides), ordered (if linkages are specified) and rooted trees.
Maximum Common Subtree Problem (MCST) • Input: Two labeled rooted trees T1 and T2. • Output: A tree which is a subtree of both tree T1 and T2 and whose number of edges is the maximum among all such possible subtrees. • Variants: Each of T1 and T2 can be ordered or unordered. Aoki, et. al. Efficient Tree-Matching Methods for Accurate Carbohydrate Database Queries. Genome Informatics 14: 134-143 (2003).
A bottom-up dynamic programming algorithm • Let {u1, …,un} and {v1, …,vm} are the sets of nodes in T1 and T2, respectively; • R[ui, vj] – the size of the maximum subtree of T1(ui) and T2(vj), the subtrees of T1 and T2 with ui and vj as roots, respectively; • Computed from leaves to roots (bottom-up) • MCST of T1 and T2 R[root(T1), root(T2)] • R[ui, ] = R[vj, ] = 0; • M(u, v) is a matching in a bipartite graph between the children of u and children of v; if both T1 and T2 are ordered trees, M(u, v) = 1. Aoki, et. al. Efficient Tree-Matching Methods for Accurate Carbohydrate Database Queries. Genome Informatics 14: 134-143 (2003). Implemented in KEGG glycan matching and many other services.
Alignment algorithm? • Complexity: unordered tree ~O(4!mn) ~ O(24mn); ordered tree ~ O(mn). Typically m, n < 25. • Extended to MCST problem in multiple trees • Is the MCST of T1, T2 and T2 is the MCST between MCST(T1, T2) and T3, where MCST(T1, T2) is the maximum subtree of T1 and T2? • Multi-MCST problem is NP-hard (Akutsu, 2002) • Reduciable from Longest Common Substring problem (LCS) • Finding substructures, motif finding problem profile models • Should we consider indels as DNA/protein alignments? • Indels is not a natural changes; but mutation might be. • Profile HMM may not be appropriate
Maximum Common Approximate Subtree Problem (MCAST) • Input: Two labeled rooted trees T1 and T2. • Output: A tree which is a k-appximatesubtreeof both tree T1 and T2 and whose number of edges is the maximum among all such possible subtrees. • T is a k-appximatesubtree of U if one of U’s subtree can be transformed to T by replacing at most k labels.
Subtree finding problem (pattern matching problem) • Input: a labeled rooted tree P and a set (database) S of labeled rooted trees. • Output: all trees in S which each has a subtree matching P. • Variants: (1) P can be ordered or unordered; (2) P must be on the root; (3) P must be on the leaves • A bottom-up DP algorithm modified from MCST algorithm; complexity O(|P|*|T|) for each T in the database.
A bottom-up dynamic programming algorithm • Let {u1, …,un} and {v1, …,vm} are the sets of nodes in P and T. • R[ui, vj] – indicator if the tree with the root of ui is a subtree of the tree with the root of vj, which is rooted by vj • Output subtree with the root of vjwhich has R[root(P), vj] = 1; • R[x, ] = R[, y] = 0. • R[x, y] = 1, if x = y and x or y is the leave of P and T, respectively. • For ordered tree, matching edges rather than nodes. • Variants: (1) leaves: R[x, y] = 1, if x = y and x and y are both leaves; (2) root: Output tree T which has R[root(P), root(T)] = 1;
Significance of matching glycans • MCST of T1 and T2 has k nodes (monosaccharides) • N(T, k): # of subtrees of T with k nodes • Can be counted by a DP algorithm (how?) • P = a-k N(T1, k) N(T2, k)
Motif retrieval from glycans • PSTMM (Probabilistic Sibling-dependent Tree Markov Model) • Learns patterns from glycan structures • Profile PSTMM • Extracts patterns (as profiles) from glycan structures • Kernel methods • Classification of glycans • Extraction of “features” to predict glycan biomarkers
Kernel method • Extracted glycan structures from CarbBank • Pre-analysis showed that the trisaccharide structure was most effective for classification • Furthermore, since the non-reducing end is usually the portion being recognized, this information was included in the kernel model
Other kernels • Q-gram distribution kernel: • Wanted to be able to analyze any data regardless of marker structure or size • Definition of q-gram: A sub-tree containing q nodes • All of the q-grams for a particular glycan were included in the kernel • Multiple kernel: • A kernel of kernels
Using a gram distribution, potential biomarkers of the appropriate size can be extracted from the data
Data mining for glycobiology • Kernels can be utilized in many ways • Feature retrieval methods for detecting putative biomarkers • Cell-specific glycan structures can be extracted • Sequences of glycan binding proteins can be included in a new kernel to predict binding domains • Many more possibilities, depending on the data