Download

1 / 33
330 likes | 641 Views
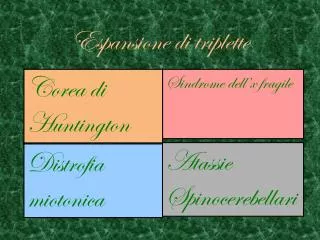
Malattie da espansione di repeats instabili. fenomeno scoperto nel 1991 (FraX, SBMA) inizialmente noto come “espansione di triplette” poi tetra- penta-nucleotidi: “ repeats instabili” Caratteristica comune: fenomeno dell’anticipazione Effetto biologico:

E N D
Malattie da espansione di repeats instabili • fenomeno scoperto nel 1991 (FraX, SBMA) • inizialmente noto come “espansione di triplette” • poi tetra- penta-nucleotidi: “repeats instabili” Caratteristica comune: fenomeno dell’anticipazione Effetto biologico: • perdita di funzione della proteina • guadagno di funzione (CAG) • effetto tossico dell’mRNA
Malattie da espansione di repeats instabili: perdita della proteina Sindrome dell’X fragile E: CCGs del 5’UTR con iper-metilazione e silenziamento del gene. Proteina ricca in serine e proline, possibile fattore trascrizionale Atassia di Friedreich: GAAs nel primo introne con ridotta espressione e inibizione dell’elongazione. Proteina “fratassina” possibile trasporto del Ferro nei mitocondri con riduzione delle ferro-proteine.
Sindrome dell’ X fragileripetizioni CGG nel 5’UTR 38 kb in Xq27.3 5 - 44 CGG con AGG ogni 9-10 triplette da 54 – 58 fino a 200 - 230 > 200
Sindrome dell’X fragile • Caratteristiche cliniche • ritardo mentale • moderato nei maschi • lieve nelle femmine • altri segni • macrocefalia, faccia allungata • oriecchie sborgenti e basse • lassità articolare • macro-orchidismo • Biologia molecolare • 99% dei casi: CGG >200 • 1% dei casi: delezioni, mut puntiformi • Prevalenza • 16-25 su 100.000 maschi
Sindrome dell’ X fragile • Maschi e femmine possono • trasmettere la pre-mutazione • Durante la meiosi materna • la pre-mutazione • può espandersi a • “mutazione”
90 CGG 28-86 CGG 26-600 CGG
Sindrome dell’ X fragile: portatori di pre-mutazione (1:300) Sindrome FXTAS (maschi) • progressiva atassia cerebellare e tremore intenzionale • perdita della memoria a breve termine, declino cognitivo • parkinsonismo, neuropatia periferica • disfunzione autonomica • debolezza muscolare dei cingoli (arti inf.) Penetranza: 17% a 60 aa 38% a 70 aa 47% a 80 aa 75% oltre gli 80 aa 2.2% delle atassie dell’età adulta 4.4% delle atassie ad esordio > 50 aa POF (femmine) - menopausa < 40aa nel 21% circa delle portatrici
Mutazioni dominanti effetto “tossico” dell’RNA messaggero • Gene FMR1 • CGG: 54-200 • POF • FXTAS • CGG > 200 • - FMR
Sindrome dell’ X fragile: Analisi in PCR Amplificazione della regione 5’UTR del gene FMR1
Sindrome dell’ X fragile: Analisi in PCR Maschio normale 29 CGGs Femmina normale 22-28 CGGs Femmina pre-M 29-58 CGGs Femmina pre-M 29-68 CGGs Femmina affetta 28 CGGs In caso di full-mutation, l’allele espanso NON è amplificabile
EcoRI EagI EcoRI 2.8 kb X attiva 5.2 kb X inattiva EcoRI EcoRI EagI Allele pre-mutato > 2.8 kb X attiva > 5.2 kb X inattiva EcoRI EcoRI EagI Allele mutato >> 5.2 kb Sindrome dell’ X fragile: Analisi in Southern blotting
Endonucleasi di restrizioneEnzimi che tagliano il DNA a doppia elica in siti specifici Blunt Ends = estremità prive di estensione
DNA a doppia elica L’ ibridazione del DNA DNA a singolo filamento Singolo filamento complementare denaturazione (soluzioni saline e/o calore) marcatura radioattiva o fluorescente SONDA
Trasferimento di acidi nucleici su filtro , ibridizzazione e auto-radiografia.
Diagnosi mediante Southern blottingdi delezioni genomiche • digestione del DNA genomico • separazione in gel di agarosio • trasferimento su nitrocellulosa • ibridazione con sonda di cDNA • analisi dei frammenti riconosciuti dalla sonda • Conclusione: • il gene testato (X linked) è assente nel figlio maschio • la madre ha solo una copia del • gene (l’intensità delle bande è • simile a quella della padre).
Diagnosi mediante Southern blottingdi delezioni genomiche. Perdita di un sito di restrizione -> banda di giunzione sonda mut WT mut (eterozigote)
Sindrome dell’ X fragile: Analisi in Southern blotting
RITARDO MENTALE • Ritardo mentale di varia entità colpisce il 3% della popolazione (si definiscono con R.M. pazienti con Q.I. inferiore a 70) • le cause eziologiche sono identificate in meno del 50% dei casi • cause esogene sono responsabili dal 18,6% al 44% dei casi di R.M. • cause genetiche sono responsabili dal 17% al 47,1% dei casi di R.M. • FRAX-A, FRX-E, anomalie cromosomiche • queste variazioni nelle percentuali possono essere dovute: • al grado di R.M. • al criterio di selezione dei pazienti esaminati • alla definizione delle diagnosi
Malattie da espansione di repeats instabili: guadagno di funzione Inclusioni citoplasmatiche , nucleari o entrambe .
Corea di Huntington • difetti di coordinazione motoria • movimenti involontari coreici (arti, faccia, tronco) • difficoltà nella programmazione delle attività • depressione, irritabilità e demenza • difficoltà nei movimenti volontari • disartria e disfagia • bradicinesia, rigidità e distonia • tentativi di suicidio (28%) • infezioni polmonari e urinarie, insufficienza cardiaca • atrofia del caudato e putamen alla TC o RMN • Esordio : 35-44 anni (raramente infantile-senile) • Decorso : 10-20 anni (progressivo)
Gene HD 4p16.3 67 esoni – mRNA di 13654 basi – 3141 aa (350kD)
Trascritto gene HD 5’ UTR Esone 1 21 CAG (10-26) Esone 2 Esone 3…..
L’età di insorgenza correla con l’entità dell’espansione Le triplette CAG si espandono preferibilmente alla meiosi paterna Range Normale: 10-26 Intermedio: 27-35 Patologico: > 36
Proteina “Huntingtina” • proteina espressa nei neuroni di diverse aree del SNC (citoplasmatica e nucleare) • perdita di funzione della proteina mutata ? • tossicità della proteina mutata ? • proteolisi • rilascio di un frammento N-term con poli-Glu • formazione di aggregati citoplasmatici e nucleari • sequestro di altre proteine e fattori di trascrizione • prococe difetto del trasporto assonale • segnale pro-apoptotico e morte cellulare
Malattie da espansione di repeats instabili: alterata funzione dell’RNA ….ed effetto biologico sconosciuto !
Il fenomeno dell’anticipazionenella distrofia miotonica Da una generazione all’altra l’età di insorgenza diminuisce e la gravità della malattia aumenta
Distrofia Miotonica: correlazione tra fenotipo e lunghezza del CTG repeat
3’UTR introne 1