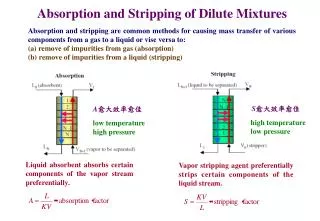
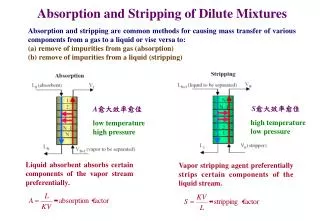
Download

1 / 36
360 likes | 476 Views
Contribution of water dimers in atmospheric absorption: methodology. Ross E. A. Kelly , Matt J. Barber, Jonathan Tennyson Department of Physics and Astronomy, University College London Gerrit C. Groenenboom, Ad van der Avoird Theoretical Chemistry Institute for Molecules and Materials,

E N D
Contribution of water dimers in atmospheric absorption: methodology Ross E. A. Kelly, Matt J. Barber, Jonathan Tennyson Department of Physics and Astronomy, University College London Gerrit C. Groenenboom, Ad van der Avoird Theoretical Chemistry Institute for Molecules and Materials, Radboud University Caviar Consortium Meeting, NPL 29th September 2010
Water Dimer Method • (1) Need to solve the nuclear motion Hamiltonian • 12D problem! Approximations required. • (2) Fully dimensional potential energy surface required • Huang, Braams and Bowman (HBB) potentials • 30-40,000 configurations sampled. • Calculated at coupled-cluster, single and double and perturbative treatment of triple excitations method. • Augmented, correlation consistent, polarized triple zeta basis set. • Polynomial fit with 5227 coefficients. HBB – X. Huang et al. J. Chem. Phys. 128, 034312 (2008). HBB2 – X. Huang et al. J. Chem. Phys.130, 144314 (2009).
(2) Fully Dimensional Water Dimer Potential Monomer corrected* HBB potential • Corrects for monomer excitation • Accurate modes for the monomer * S. V. Shirin et al., J. Chem. Phys. 128, 224306 (2008). R.E.A. Kelly, J. Tennyson, G C. Groenenboom, A. Van der Avoird, JQRST, 111, 1043 (2010).
(1) Fully Dimensional (12D) Solution • Only useful for 12D vibrational ground state. • Diffusion Monte Carlo may be used • Start with a number of walkers • Allow them to follow a random walk in space • Propagate in imaginary time • Decide whether to replicate or destroy the walker
Solving the 6D intermolecular problem • Brocks et al. Hamiltonian* • Monomers fixed in • Equilibrium geometry, or • Vibrational ground state geometry * G. Brocks et al. Mol. Phys. 50, 1025 (1983).
Acceptor Twist (AT) Acceptor Wag (AW) Donor Torsion (DT) Solving the 6D intermolecular problem Solving the 6D intermolecular problem In Plane Bend (IPB) Out-of-Plane Bend (OPB) Stretch Generated by Matt Hodges and Anthony Stone. C. Millot et al.J. Phys. Chem. A 1998,102, 754. http://www-stone.ch.cam.ac.uk/research/water.dimer/modes.html
Tunneling Splittings 2 2 6 6 4 3 1 1 5 5 3 4 1 1 2 6 2 6 3 4 5 5 4 3 1 2 1 2 6 6 3 3 5 5 4 4 • Isomorphic to D4h withIrreducible Elements: A1+, A2+, A1-, A2-, B1+, B2+, B1-, B2-, E+, E- -> Water Dimer Spectroscopic Labels 2 2 1 1 6 6 4 4 5 5 3 3
Tunneling Splittings • Very good agreement with: • Ground State Tunnelling splittings • Rotational Constants • Not so good agreement with: • Acceptor Tunnelling
Solving the 6D intermolecular problem Brocks et al. Hamiltonian* Monomers fixed in Equilibrium geometry, or Vibrational ground state geometry Is there another way to help us probe the 12D problem? * G. Brocks et al. Mol. Phys. 50, 1025 (1983).
Adiabatic Separation • Approximate separation between monomer and dimer modes • Separate intermolecular and intramolecular modes. mD – water donor vibrational wavefunction mA – water acceptor vibrational wavefunction d – dimer VRT wavefunction
Solving the 6D intermolecular problem • Now we can vibrationally average the potential • Input for 6D calculations • donor acceptor • State m State n • How well does it perform for |0 0> calculations
Vibrational Averaging • In cm-1 • Red – ab initio potential • Black – experimental • GS – ground state • DT – donor torsion • AW – acceptor wag • AT – acceptor twist • DT2 – donor torsion overtone R.E.A. Kelly, J. Tennyson, G C. Groenenboom, A. Van der Avoird, JQRST, 111, 1043 (2010).
Vibrational Averaging: 6D Costs! • Computation: • typical number of DVR points with different Morse Parameters: • {9,9,24} gives 1,080 points for monomer • 1,0802 = 1,166,400 points for both monomers • 1,166,400 x 2,894,301 intermolecular points = 3,374,862,926,400 points • Same monomer wavefunctions for all grid points • Distributed computing: Condor 1000 computers, 10 days But we have a way to probe high frequency dimer spectra
Full model for high frequency absorption Approximate separation between monomer and dimer modes Franck-Condon approximation for vibrational fine structure Rotational band model
Adiabatic Separation • Approximate separation between monomer and dimer modes • Separate intermolecular and intramolecular modes. mD – water donor vibrational wavefunction mA – water acceptor vibrational wavefunction d – dimer VRT wavefunction
Model for high frequency absorption Approximate separation between monomer and dimer modes Franck-Condon approximation for vibrational fine structure Rotational band model
Franck-Condon Approx for overtone spectra Assume monomer m1 excited, m2 frozen m2i = m2f I a (2) Franck-Condon factor (square of overlap integral): Gives dimer vibrational fine structure (1) Monomer vibrational band Intensity
Allowed Transitions in our Model Assume excitation localised on one monomer 2. Excited acceptor 1. Excited donor All transitions from ground monomer vibrational states
Franck-Condon factors • Overlap between dimer states on adiabatic potential energy surfaces for water monomer initial and final states • Need the dimer states (based on this model).
Transitions: Example Donor – Vibrational ground state (VGS) Acceptor – VGS Donor –VGS Acceptor – bend Acceptor Twist (AT) Acceptor Wag (AW) Donor Torsion (DT) Ground State (GS) Acceptor Twist (AT) Acceptor Wag (AW) Donor Torsion (DT) Ground State (GS)
Transitions: Example Donor – Vibrational ground state (VGS) Acceptor – VGS Donor –VGS Acceptor – bend Acceptor Twist (AT) Acceptor Wag (AW) Donor Torsion (DT) Ground State (GS) Acceptor Twist (AT) Acceptor Wag (AW) Donor Torsion (DT) Ground State (GS)
Transitions: Example Donor – Vibrational ground state (VGS) Acceptor – VGS Donor –VGS Acceptor – bend Acceptor Twist (AT) Acceptor Wag (AW) Donor Torsion (DT) Ground State (GS) Acceptor Twist (AT) Acceptor Wag (AW) Donor Torsion (DT) Ground State (GS)
Outline of full problem • Calculate states for donor • Calculate states for acceptor • Vibrationally average potential for each state-state combination • Really only |0j> and |i0> • Need to ultimately solve (6D problem) • H=K+Veff • Veff sampled on a 6D grid
(a) 6D averaging: (b) 3D+3D averaging: 1 C Leforestier et al,J Chem Phys, 117, 8710 (2002) 2 R. E. A. Kelly et al. To submit shortly. Averaging Techniques
Averaging Techniques • Form of the wavefunction: • (I) Uncoupled free monomer • (II) Uncoupled perturbed (fixed) monomer • R. E. A. Kelly et al. To submit shortly.
Problems with Fixed Wavefunction approach (uncoupled methods) • Donor bend • (Donor) Free OH stretch • (Donor) Bound OH stretch • (Donor) Free OH stretch • (Donor) Bound OH stretch
Averaging Techniques • Form of the wavefunction: • (I) Uncoupled free monomer • (II) Uncoupled perturbed (fixed) monomer • (III) Coupled Adiabatic • R. E. A. Kelly et al. To submit shortly.
Averaging Techniques • Form of the wavefunction: • (I) Uncoupled free monomer • (II) Uncoupled perturbed (fixed) monomer • (III) Coupled Adiabatic • R. E. A. Kelly et al. To submit shortly.
Averaging Techniques • Form of the wavefunction: • (I) Uncoupled free monomer • (II) Uncoupled perturbed (fixed) monomer • (III) Coupled Adiabatic • R. E. A. Kelly et al. To submit shortly.
Averaging Techniques • Form of the wavefunction*: • (I) Uncoupled free monomer • (II) Uncoupled perturbed monomer • (III) Coupled Adiabatic • Coupled Adiabatic methods are the most suitable • Requires wavefunction calculations at each intermolecular grid point! 2,893,401 * 2 DVR3D calculations! • So we use cheaper (3+3)D averaging technique. • Still costs! 500-700 CPUs for 3-4 weeks. • *R. E. A. Kelly et al. To submit shortly.
Calculating dimer spectra with FC approach • Solved for monomers • Coupled adiabatic appoach • Vibrationally averaged potential for donor-acceptor state-state combinations |0j> and |i0> • Input for 6D intermolecular problem • Now we can solve 6Dintermolecular problem • Obtain vibrational fine structure
Solving the 6D intermolecular problem:Allowed permutations 2 2 6 6 4 3 1 1 5 5 3 4 1 1 2 6 2 6 3 4 5 5 4 3 1 2 1 2 6 6 3 3 5 5 4 4 2 2 1 1 6 6 4 4 5 5 3 3
Solving the 6D intermolecular problem:Allowed permutations for excited monomers 2 2 6 6 4 3 1 1 5 5 3 4 • G16 Symmetry of Hamiltonian for GS monomers • > replaced with G4 • Greatly increases computational requirements • Reduced angular basis • Small radial basis • 320 diagonalizations for 0-10,000 cm-1 • Each at 16GB • 8 states per symmetry block • Leading to 20,480 transitions
Strongest absorption on bend – difficult to distinguish from monomer features More structure between 6000-9000 cm-1 Full Vibrational Stick Spectra
Model for high frequency absorption Approximate separation between monomer and dimer modes Franck-Condon approximation for vibrational fine structure Rotational band model – Matt will discuss this
Conclusions • We have a new model to probe near IR and visible regions of the water dimer spectra. • With first vibrational fine structure reported. • Spectra for up to 10,000 cm-1 produced. • Much better agreement with experimental and theoretical work than our previous calculations. • We have finished new averaging calculations which will allow us to probe spectra up to 18,000 cm-1 • And all states up to dissociation to be calculated. • Only 8 states per symmetry here (32 states per state-state job) • up to 800, or 3200 per job.