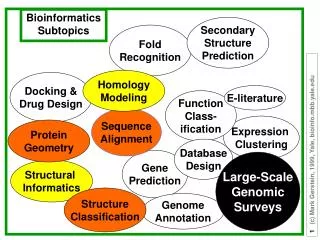
Fold Recognition
Fold Recognition. Ole Lund, Associate professor, CBS. Fold recognition. Find template for modeling 1st step in comparative modeling Can be used to predict function. Template identification. Search with sequence Blast against proteins with known structure Psi-Blast against all proteins


Fold Recognition
E N D
Presentation Transcript
Fold Recognition Ole Lund, Associate professor, CBS
Fold recognition • Find template for modeling • 1st step in comparative modeling • Can be used to predict function OL
Template identification • Search with sequence • Blast against proteins with known structure • Psi-Blast against all proteins • Fold recognition methods • Use biological information • Functional annotation in databases • Active site/motifs OL
Blast derivatives: PDB-BLAST • Procedure • Build sequence profile by iterative PSI-BLAST search against a sequence database • Use profile to search database of proteins with known structure • Advantage • Makes sure hid to protein with known structure is not hidden behind a lot of hits to other proteins OL
BLAST derivatives: Transitive BLAST • Procedure • Find homologues to query (your) sequence • Find homologues to these homologues • Etc. • Can be implemented with e.g. BLAST or PSI-BLAST • Also known as Intermediate Sequence Search (ISS) OL
CASP • CASP • Critical Assessment of Structure Predictions • Every second year • Sequences from about-to-be-solved-structures are given to groups who submit their predictions before the structure is published • Modelers make prediction • Meeting in Asilomar where correct answers are revealed OL
Target difficulty • CM: Comparative (homology) modeling • CM/FR: not PSI-BLAST (but ISS) findable • FR(H): Homologous fold recognition • FR(A): Analogous fold recognition • NF/FR: Partly New fold • NF: New Fold (used to be called Ab Initio -from first principles- prediction) OL
Successful fold recognition groups at CASP5 • 3D-Jury (Leszek Rychlewski) • 3D-CAM (Krzysztof Ginalski) • Template recombination (Paul Bates) • HMAP (Barry Honig) • PROSPECT (Ying Xu) • ATOME (Gilles Labesse) OL
Abstract Barry Honig • Sequence&structure profile-profile based alignment • Database of template profiles • Multiple structure alignment • Sequence based profiles • Position specific gap penalties derived from secondary structure • Calibration to estimate statistical significance • Query profile • Sequence based profile • Predicted secondary structure (consensus between PSI-PRED,PHD,JNET) OL
Abstract Ying Xu • PROSPECT:optimal alignments for a given energy function with any combination of the following terms: • mutation energy (including position-specific score matrix derived from multiple-sequence alignments), • singleton energy (including matching scores to the predicted secondary structures), • pairwise contact potential • alignment gap penalties. OL
3D-Jury (Rychlewski) • Inspired by Ab initio modeling methods • Average of frequently obtained low energy structures is often closer to the native structure than the lowest energy structure • Find most abundant high scoring models • Use output from a set of servers • Superimpose all pairs of structures • Similarity score Sij = # of Ca pairs within 3.5Å (if #>40;else Sij=0) • 3D-Jury score = SiSij/(N+1) • Similar methods developed by A Elofsson (Pcons) and D Fischer (3D shotgun) OL Rychlewski.doc
Ginalski.doc 3D-CAM (Krzysztof Ginalski) • 3D-Consensus Alignment Method • Structural alignment for all members of fold from FSSP • Conservation of specific residues and contacts • responsible for maintaining tertiary structure • critical for substrate binding and/or catalysis • Find homologues with iterative PSI-BLAST • Align with ClustalW – identify conserved residues • Structural integrity of alignments • Manual realignment • Fold recognition for homologues • Modelling • Verification • Visually • Computationally (Verify3D, ProsaII, WHAT_CHECK) OL
Abstract Paul A Bates - In Silico Recombination of Templates, Alignments and Models • Problems • Models rarely better than templates • Manual intervention have marginal effect • Possible solution • Recombination of models OL
Abstract Paul A Bates – Modelling Procedure • Define domains • Make models (FAMS/Pmodeller/EsyPred3D) • Manual inspection/correction of alignments • Alignment of annotated residues (PFAM) • Preferably use alignment with >2 bits/aa • Select pair of models • Superimpose • Crossover or mutate (average coordinates) • Select best proportion • Contact pair potentials • Solvation energies (calculated from solvent accessible area) • Convergence • Minimization and final refinements OL
LiveBench • The Live Bench Project is a continuous benchmarking program. Every week sequences of newly released PDB proteins are being submitted to participating fold recognition servers. The results are collected and continuous evaluated using automated model assessment programs. A summary of the results is produced after several months of data collection. The servers must delay the updating of their structural template libraries by one week to participate. OL
Meta Server OL
http://bioinfo.pl/meta/target.pl?id=7296 Meta Server OL
Score # wrong # correct OL
Best servers? • FFA3 • 3DS5 • INBG • SHUM • 3DPS • 3DS3 • FUG3 • SHGU • FUG2 • PCO2 • PRO2 • MGTH • SFPP • PMO3 OL
Links to fold recognition servers • Databases of links • http://bioinfo.pl/meta/servers.html • http://mmtsb.scripps.edu/cgi-bin/renderrelres?protmodel • Meta server • http://bioinfo.pl/meta/ (Example: http://bioinfo.pl/meta/target.pl?id=7296 ) • 3DPSSM – good graphical output • http://www.sbg.bio.ic.ac.uk/servers/3dpssm/ • GenTHREADER • http://bioinf.cs.ucl.ac.uk/psipred/ • FUGUE2 • http://www-cryst.bioc.cam.ac.uk/~fugue/prfsearch.html • SAM • http://www.cse.ucsc.edu/research/compbio/HMM-apps/T99-query.html • FOLD • http://fold.doe-mbi.ucla.edu/ • FFAS/PDBBLAST • http://bioinformatics.burnham-inst.org/ OL