Enzyme Kinetics
Kinetic concepts Kinetics is the study of the rates of chemical reactions. Unlike Thermodynamics which tells us if a reaction can occur, kinetics provides information on the rate and mechanism of the reaction. Enzyme Kinetics. Chemical kinetics.


Enzyme Kinetics
E N D
Presentation Transcript
Kinetic concepts Kinetics is the study of the rates of chemical reactions. Unlike Thermodynamics which tells us if a reaction can occur, kinetics provides information on the rate and mechanism of the reaction. Enzyme Kinetics
Chemical kinetics • It is important to recognize that, although a reaction may proceed with the stoichiometry, A P • The reaction may proceed via a series of elementary reactions such as A I1 I2 P • A description of the elementary reactions and intermediates is essential for defining the mechanism of a reaction. • This is mostly basic chemical kinetics with a biological twist to account for the problems of enzymes.
-d[A] d[P] = = k[A] = v dt dt Fundamental kinetic concepts rate constant k A P Rate of disappearance of reactant Rate of appearance of product The instantaneous rate, or velocity (v), of the reaction at any time is reflected in the disappearance of reactant and/or in the appearance of product. The rate is directly proportional to the concentration of the reactant, A. This proportionality is reflected in the rate constant, k. The above reaction is an example of a first-order overall reaction
-d[A] d[P] = = k[A]2 = v dt dt Fundamental kinetic concepts A + A P k Rate Law k A + B P -d[A] -d[B] d[P] = = = k[A][B] = v dt dt dt Rate Law The above reactions are both second-order overall reactions. (The overall order of a reaction comes from adding together the exponents in the rate law) Note: Rate orders can NOT be deduced from the balanced chemical equation. They can only be determined experimentally.
Units for rate constants • The units for rate constants differ depending on whether they are first or second order: • The first order differential rate equation has units M·s-1, therefore k must have units s-1. • For second order rate equations (M·s-1) = k(M2). In order for the units to balance, the units for k must be (M-1s-1). • The order of a reaction can be determined by following the rate as a function of time, and fitting to either a first or second order equation. v (Ms-1) = k (s-1)[A] (M) v (Ms-1) = k (s-1M-1)[A] (M) [B] (M)
–d[A] d[P] = = k[A] dt dt t ∫ 0 First order rate equation • We want to rearrange the equation describing the instantaneous reaction velocity into a more useful form, where the change in [A] is expressed as a function of time: • Rearranging gives: • Which may be integrated from [A]o the initial concentration to [A] at time t: • Which results in: 1 d[A] = –kdt [A] [A] 1 ∫ d[A] = –k dt [A] [A]0 For a first-order reaction, there exists a linear relationship between ln[A] and t
First order rate curves • The half-time or half-life, t1/2 is constant for a first order reaction. From the rate equation: The half-life for a first-order reaction is independent of the initial concentration of the reactant.
Second order rate equations for single reactant systems • In a second order reaction for the type 2A P the variation of [A] is quite different from the first order reaction. Rearranging and integrating • Gives • So that • For a second-order reaction, there exists a linear relationship between 1/[A] and t. Thus, it is trivial to distinguish between first and second order reactions by the nature of the rate vs. concentration dependence (see the plot on the following page)
Comparison of rate curves • The t1/2 for a second order reaction is (try deriving this expression yourself) • t1/2 for a second-order reaction is dependent on the initial concentration of reactant and, thus, differs from the first order reaction.
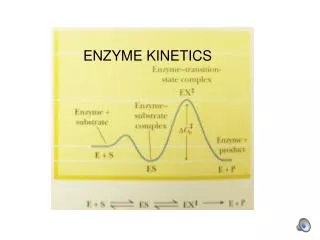
Enzyme kinetics: The rapid-equilibrium approach(The Henri-Michaelis-Menten Equation) • A general scheme for a simple enzyme-catalyzed reaction which converts a single substrate into a single product is: E + S ES EP E + P • This kinetic scheme is simplified significantly when the reaction proceeds at “initial velocity”. i.e. at the onset of the reaction, [S] = 100% while [P] = 0%. While [P] remains very low, the back reaction is negligible and the above scheme may be simplified to: E + S ES E + P • The assay of an enzyme under initial velocity conditions is, therefore, an important consideration in the practical design of enzyme assays. We will revisit this later. k1 k3 k2 k-1 k-3 k-2 k1 kcat k-1
-d[S] d[P] vi= = = kcat[ES] dt dt Derivation of the Henri-Michaelis-Menten equation • The rate of the reaction is measured by the appearance of product (or the disappearance of substrate). The overall rate of the reaction is governed by the rate of conversion of the final intermediate (in this case, ES) into free enzyme (E) and free product (P). Thus, the rate of the above reaction is given by: [eq. 1] • The preceding equation is not particularly useful. Since ES is an intermediate in the reaction, its concentration at any given time is unknown and it is not practical to directly follow its conversion into E + P. It would be much more useful to express the rate equation in terms of the total enzyme concentration ([E]t) and the initial substrate concentration ([S]), both of which are known. • The derivation of such an expression requires certain assumptions. The simplest derivation uses the “Rapid-Equilibrium assumption”.
Derivation of the Henri-Michaelis-Menten equation k1 [E][S] k-1 • Assume Rapid Equilibrium (k-1 >> kcat): E + S ES KS = = [eq. 2] • The total enzyme concentration ([E]t) is the sum of all enzyme species: [E]t = [E] + [ES] [eq. 3] • Divide [eq. 1] by [eq. 3] vikcat[ES] [E]t [E] + [ES] and, from rearranging [eq. 2], [ES] k1 k-1 Note: KS is a dissociationconstant = [eq. 4] [E][S] [[ES] = [eq. 5] KS
Vmax[S] vi = [S] + KS Derivation of the Henri-Michaelis-Menten equation • Now, substitute [eq. 5] into [eq. 4]: vikcat [E][S] KS [E]t [E] + [E][S] KS vikcat [S] KS [E]t 1 + [S] KS (kcat [E]t)[S] [S] + KS = KS = multiply the numerator and denominator by KS KS Enzymologists define: vi = (kcat [E]t)= Vmax where KS is a true dissociation constant
The Briggs-Haldane steady state approach • The Henri-Michaelis-Menten equation was originally developed by Victor Henri (1903) and later confirmed by Leonor Michaelis and Maud Menten (1913). • The assumption of rapid equilibrium in the derivation of the Henri-Michaelis-Menten equation requires that the rate of dissociation of the ES complex (k-1) far exceed the rate of conversion of the ES complex into E + P (kcat). Unfortunately, this assumption is invalid for many (if not most) enzymes. • In 1925, Briggs & Haldane developed an initial velocity rate equation that did not require the assumption of rapid equilibrium. Rather, the Briggs-Haldane approach was to assume a steady-state for the ES complex.
The Briggs-Haldane steady state approach Steady state assumption: • When [S] >> [E], the level of [ES] stays constant after an initial burst phase. i.e. d[ES]/dt ≈ 0 Note: The accompanying figure is somewhat deceptive. In fact, steady state is reached very quickly. The initial phase of an enzyme-catalyzed reaction, prior to the onset of steady state, can only be followed using specialized equipment in combination with rapid sample mixing techniques. The kinetics are much more complex but they can yield important information about the individual kinetic steps in an enzyme-catalyzed reaction. This type of kinetics is referred to as “pre-steady state”
Derivation of the Briggs-Haldane equation • As before, this derivation deals only with initial velocity kinetics. We can treat the reverse reaction as neglible and simplify the scheme to: • E + S ES E + P • The overall rate of production of ES is the sum of the elementary reaction rates leading to its appearance minus the sum of those leading to its disappearance. k1 k2 k-1 (Note that k2 is analogous to kcat) d[ES] = k1[E][S] – k-1[ES] – k2[ES] = 0 dt k1[E][S] Rearranging: [ES] = [eq. 6] k-1 + k2
-d[S] d[P] vi= = = k2[ES] dt dt Derivation of the Briggs-Haldane equation • As before, and vik2[ES] [E]t [E] + [ES] = Substituting in [eq. 6], we get: vik2k1[E][S] (k-1 + k2) [E]t [E] + [E][S]k1 (k-1 + k2) = k2[E]t[S] vi = [eq. 7] k-1 + k2 + [S] k1
k-1 + k2 k1 Km = Vmax[S] vi = [S] + Km Derivation of the Briggs-Haldane equation The Michaelis constant, Km, has units of M and is defined as: and (k2 [E]t)= Vmax Substituting these into [eq. 7] gives the final form of the Briggs-Haldane equation: Some special cases: When [S] >> Km, vi = Vmax (i.e. velocity is independent of [S]; The enzyme is said to be “at saturation”) When [S] << Km, vi = (Vmax/Km)[S] (i.e. the velocity is linear with respect to [S])
The meaning of Km Substitution of =Vmax/2 into the Briggs-Haldane equation shows that: The value of Km does NOT necessarily give a measure of the affinity of the enzyme for the substrate. When k2is small relative to k-1, Km approximates to the dissociation constant of the ES complex, KS. Since KS is a true dissociation constant, only KS gives a true measure of the enzyme-substrate binding affinity. Km ≥ KS
The meaning of Vmax , kcat , and Specific Activity Vmax(Ms-1) is the maximal velocity of an enzyme-catalyzed reaction. The maximal velocity is reached when the substrate is saturating. Vmax is dependent on [E]t. kcat (s-1) is the catalytic constant, also called the “turnover number”. It is a pseudo-first order rate constant and is independent of the total enzyme concentration. (kcat [E]t)= Vmax Specific Activity (U/mg total protein) is often used to characterize enzyme activity when the enzyme solution is impure. It is, most always, a quick but less accurate means of kinetically characterizing an enzyme. 1 International Unit (1 U) is the amount of enzyme which catalyzes the formation of 1 mmole of product per minute under defined conditions. i.e. 1 U = 1 mmole/min
k1 k3 k2 E + S ES EP E + P k-1 k-2 The kinetic significance of kcat kcat, a pseudo-first order rate constant, includes the individual rate constants for all steps leading from the ES complex to product release. For example, in a more complex kinetic scheme such as: kcat ES E + P It can be shown that kcat is comprised of all the individual rate constants between ES and E + P (k2, k-2 and k3): k2k3 kcat = (k2 + k-2 + k3)
The kinetic significance of kcat/Km When the substrate concentration is very low ([S] << Km): vi = (Vmax/Km)[S] Recall that Vmax = kcat[E]t So, vi = (kcat/Km)[S][E]t Also, when [S] is very low, very little of the total enzyme species will be tied up in the ES complex (or any other intermediate complex). [E]t = [E] + [ES] But at low substrate concentrations, [E]t ≈ [E] (because [ES] ≈ 0) thus, vi = (kcat/Km)[S][E] when [S] << Km kcat/Km (catalytic efficiency) is a second-order rate constant that describes the conversion of free E and free S into E + P. The rate at low [S] is directly proportional to the rate of enzyme-substrate encounter.
Catalytic Perfection = k1 when k2 >> k-1 • How quickly can an enzyme convert substrate into product following enzyme-substrate encounter? This depends on the rates of the individual steps in the reaction. The rate is maximal when k2>>k-1 which means that the reaction proceeds whenever a collision occurs. • Many enzymes in metabolic pathways have evolved to function at substrate concentrations less than Km to optimally and efficiently turnover metabolic intermediates. • Some enzymes are so incredibly efficient that they instantaneously convert S into P following enzyme-substrate encounter. The reaction rate for these enzymes is limited only by the rate of diffusion (~ 108– 109 M-1s-1). These enzymes are said to have reached evolutionary perfection.
Examples of rate constants • There is a wide variation in kinetic parameters reflecting the interplay between KM and kcat. Because of the central role of the Enzyme·Substrate complex, there is also large variability depending on the nature of the substrate.
Analysis of Kinetic Data • The rate equations are non-linear, so it is convenient to reformulate the equation to give a linear relationship. The most common linearization is the Lineweaver-Burk or double reciprocal plot, obtained from the reciprocal of the Michaelis-Menten equation. • DISADVANTAGE: the data usually involve large ratios of KM so the data is crowded. At low [S] values the errors are often large. • There are several other linear forms of the Michaelis-Menten equation. However, most data is now easily analyzed directly on computer by a direct non-linear regression fit to the Michaelis-Menten equation.
Michaelis-Menten Kinetics Km is the substrate concentration at which the rate of the reaction is half the maximum rate (Vmax)