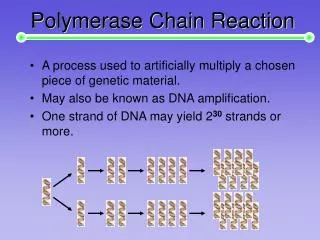
Polymerase Chain Reaction (basic principal)
Polymerase Chain Reaction (basic principal). Polymerase Chain Reaction (PCR). Developed by Kary Mullis and described in 1985 (Saiki et al., 1985) Mimics repeated cycles of DNA replication to amplify specific sequences of DNA Subsequently modified to use RNA as the template for replication.


Polymerase Chain Reaction (basic principal)
E N D
Presentation Transcript
Polymerase Chain Reaction (PCR) • Developed by Kary Mullis and described in 1985 (Saiki et al., 1985) • Mimics repeated cycles of DNA replication to amplify specific sequences of DNA • Subsequently modified to use RNA as the template for replication
Within a dividing cell, DNA replication involves series of enzymes mediated reaction : for unwind double helix DNA (helicases) For synthesis of primers (a RNA polymerase / primase) For DNA synthesis (DNA -polymerase) In PCR : High temperature to denature dsDNA ssDNA Primers: synthetically pre-made DNA polymerase : thermostable Taq – DNA polymerase WHAT IS PCR ?
PCR PCR is a technique that takes a specific sequence of DNA of small small amounts and amplifies it to be used for further works The Polymerase Chain Reaction (PCR) provides an extremely sensitive means of amplifying relatively large quantities of DNA First described in 1985, Nobel Prize for Kary Mullis in 1993 The technique was made possible by the discovery of Taq polymerase, the DNA polymerase that is used by the bacterium Thermus aquaticus that was discovered in hot springs
What is PCR ? • Method for exponential amplification of DNA or RNA sequences • Basic requirements • template DNA or RNA • 2 oligonucleotide primers complementary to different regions of the template • heat stable DNA polymerase • 4 nucleotides and appropriate buffer
DNA nucleotides, the building blocks for the new DNA Template DNA, the DNA sequence that you want to amplify Primers, oligonucleotides that are complementary to a shortregion on either side of the template DNA DNA polymerase, a heat stable enzyme that drives, orcatalyzes, the synthesis of new DNA The targets in PCR are the sequences of DNA on each end of the region of interest, which can be a complete gene or small sequence. The primary materials, or reagents, used in PCR are:
The Cycling Reactions : There are three major steps in a PCR (denaturation, annealing & extension), which are repeated for 20 to 40 cycles. This is done on an automated Thermo Cycler, which can heat and cool the reaction tubes in a very short time. Denaturation at around 94°C : During the denaturation, the double strand melts open to single stranded DNA, all enzymatic reactions stop (for example the extension from a previous cycle). Annealing at around 54°C : Hydrogen bonds are constantly formed and broken between the single stranded primer and the single stranded template. If the primers exactly fit the template, the hydrogen bonds are so strong that the primer stays attached Extension at around 72°C : The bases (complementary to the template) are coupled to the primer on the 3' side (the polymerase adds dNTP's from 5' to 3', reading the template from 3' to 5' side, bases are added complementary to the template)
PCR Every cycle results in a doubling of the number of strands DNA present After the first few cycles, most of the product DNA strands made are the same length as the distance between the primers The result is a dramatic amplification of a the DNA that exists between the primers. The amount of amplification is 2 raised to the n power; n represents the number of cycles that are performed. After 20 cycles, this would give approximately 1 million fold amplification. After 40 cycles the amplification would be 1 x 1012
Critical Factors for Successful PCR • Equipment • Thermal cycling profile & cycle number • Enzyme concentration • MgCl2Concentration • dNTPs Conc. • Primer sequences • Template
.Equipment : • Type of thermocycler : • - accurately & reproducibly 3 PCR incubation temperature • - Ramp • - cycle between temperatures repeatedly & reproducibly • b. Type of reaction tubes: affect heat transfer • - fit precisely into wells • - thin walls
PCR Optimization • Yield • Specificity • Fidelity • Size of product • Robustness of reaction
Yield • Optimum yield for a given number of cycles can be approximated using the formula: 2n x initial number of copies. n = # of cycles • There is an exponential phase of amplification in PCR until the number of product copies reaches approximately 1012. After this point the accumulation of product amplification generally drops off dramatically and the product stops accumulating exponentially. The number of cycles needed to reach this point can be approximated with the formula:Nf = No(1 + Y)nWhere Nf is the final number of the double stranded target sequence No is the original number of target copies Y is the efficiency per cycle of the polymerase n is the number of cycles
Factors That Influence Specificity • Primer design • Enzyme/reaction set-up • Reaction optimization • Magnesium concentration • Buffer formulation • Annealing temperature
Factors That Influence Fidelity • Number of cycles • Magnesium concentration • Nucleotide concentration and balance • Enzyme choice
Cycle Number • Standard Range: 20-30 cycles The plateau effect encourages nonspecific amplification and so increasing the number of cycles does not increase specificity or efficiency of your PCR. • Improve specificity: Reduce number of cycles. Reduce cycle segment lengths. • Improve efficiency: For amplifying large fragments (> 1 kb) increase the duration of each thermal step.
The “Plateau Effect” The plateau effect:attenuation in the exponential rate of product accumulation in late stages of a PCR, when product reaches 0.3-1.0 nM.This may be caused by degradation of reactants (dNTPs, enzyme); reactant depletion (primers, dNTPs - former a problem with short products, latter for long products); end-product inhibition (pyrophosphate formation); competition for reactants by non-specific products; competition for primer binding by re-annealing of concentrated (10nM) product.
Several conditions can effect the plateau: • The utilization of substrates, either primers or dNTPs. • The stability of the reactants. • End product inhibition. • Competition for reactants by nonspecific products or primer-dimers. • Reannealing of product at higher concentrations which prevents the extension process. • Incomplete denaturation at higher product concentration.
Perhaps the most critical parameter for successful PCR is the design of primers Primer design General notes on primer design in PCR Primer selection Critical variables are: - primer length - melting temperature (Tm) - specificity - complementary primer sequences - G/C content - 3’-end sequence
Primer length specificity and the temperature of annealing are at least partly dependent on primer length oligonucleotides between 20 and 30 (50) bases are highly sequence specific primer length is proportional to annealing efficiency: in general, the longer theprimer, the more inefficient the annealing the primers should not be too short as specificity decreases Primer design
Primer design Specificity Primer specificity is at least partly dependent on primer length: there are many more unique 24 base oligos than there are 15 base pair oligos Probability that a sequence of length n will occur randomly in a sequence of length m is: Example: the mtDNA genome has about 20,000 bases, the probability of randomly finding sequences of length n is: n Pn 5 19.52 10 1.91 x 10-2 15 1.86 x 10-5 P = (m – n +1) x (¼)n
Primer design • Complementary primer sequences • primers need to be designed with absolutely no intra-primer homology beyond 3 base pairs. If a primer has such a region of self-homology, “snap back” can occur • - another related danger is inter-primer homology: partial homology in the middle regions of two primers can interfere with hybridization. If the homology should occur at the 3' end of either primer, primer dimer formation will occur • G/C content • ideally a primer should have a near random mix of nucleotides, a 50% GC content • there should be no PolyG or PolyC stretches that can promote non-specific annealing 3’-end sequence - the 3' terminal position in PCR primers is essential for the control of mis-priming - inclusion of a G or C residue at the 3' end of primers helps to ensure correct binding (stronger hydrogen bonding of G/C residues)
Primer design Melting temperature (Tm) • the goal should be to design a primer with an annealing temperature of at least 50°C • the relationship between annealing temperature and melting temperature is one of the “Black Boxes” of PCR • a general rule-of-thumb is to use an annealing temperature that is 5°C lower than the melting temperature • the melting temperatures of oligos are most accurately calculated using nearest neighbor thermodynamic calculations with the formula: • Tm = H [S+ R ln (c/4)] –273.15 °C + 16.6 log 10 [K+] • (H is the enthalpy, S is the entropy for helix formation, R is the molar gas constant and c is the concentration of primer) • a good working approximation of this value can be calculated using the Wallace formula: • Tm = 4x (#C+#G) + 2x (#A+#T) °C • both of the primers should be designed such that they have similar melting temperatures. If primers are mismatched in terms of Tm, amplification will be less efficient or may not work: the primer with the higher Tm will mis-prime at lower temperatures; the primer with the lower Tm may not work at higher temperatures.
TGCT AGTTG A Uniqueness There shall be one and only one target site in the template DNA where the primer binds, which means the primer sequence shall be unique in the template DNA. There shall be no annealing site in possible contaminant sources, such as human, rat, mouse, etc. (BLAST search against corresponding genome) Template DNA5’...TCAACTTAGCATGATCGGGTA...GTAGCAGTTGACTGTACAACTCAGCAA...3’ TGCTAAGTTG CAGTCAACTGCTAC NOT UNIQUE! Primer candidate 1 5’-TGCTAAGTTG-3’ UNIQUE! Primer candidate 2 5’-CAGTCAACTGCTAC-3’
Length Primer length has effects on uniqueness and melting/annealing temperature. Roughly speaking, the longer the primer, the more chance that it’s unique; the longer the primer, the higher melting/annealing temperature. Generally speaking, the length of primer has to be at least 15 bases to ensure uniqueness. Usually, we pick primers of 17-28 bases long. This range varies based on if you can find unique primers with appropriate annealing temperature within this range.
GCCCG CAT T T AT GC Base Composition • Base composition affects hybridization specificity and melting/annealing temperature. • Random base composition is preferred. We shall avoid long (A+T) and (G+C) rich region if possible. Template DNA5’...TCAACTTAGCATGATCGGGCA...AAGATGCACGGGCCTGTACACAA...3’ TGCCCGATCATGCT • Usually, average (G+C) content around 50-60% will give us the right melting/annealing temperature for ordinary PCR reactions, and will give appropriate hybridization stability.
Internal Structure If primers can anneal to themselves, or anneal to each other rather than anneal to the template, the PCR efficiency will be decreased dramatically. They shall be avoided. However, sometimes these 2 structures are harmless when the annealing temperature does not allow them to take form. For example, some dimers or hairpins form at 30 C while during PCR cycle, the lowest temperature only drops to 60 C.
Primer Pair Matching Primers work in pairs – forward primer and reverse primer. Since they are used in the same PCR reaction, it shall be ensured that the PCR condition is suitable for both of them. One critical feature is their annealing temperatures, which shall be compatible with each other. The maximum difference allowed is 3 C. The closer their Tanneal are, the better.
Summary ~ when is a “primer” a primer? 5’ 3’ 5’ 3’ 5’ 3’ 3’ 5’
Summary ~ Primer Design Criteria • Uniqueness: ensure correct priming site; • Length: 17-28 bases.This range varies; • Base composition: average (G+C) content around 50-60%; avoid long (A+T) and (G+C) rich region if possible; • Optimize base pairing: it’s critical that the stability at 5’ end be high and the stability at 3’ end be relatively low to minimize false priming. • Melting Tm between 55-80 C are preferred; • Assure that primers at a set have annealing Tm within 2 – 3 C of each other. • Minimize internal secondary structure: hairpins and dimmers shall be avoided.
MgCl2 concentration : • Opt. 0.5-5.5mM, increase increase PCR product but decrease specificity • Mg‡ : - influences enzyme activity • - increase Tm dsDNA & forms soluble complexes with dNTPs to produce actual substrate that the polymerase recoqnize • - Conc. Mg‡ depends on conc.compound that binds the ion such as dNTPs, Ppi, EDTA
Magnesium Concentration • Standard Range: 0.5 - 10 mM Free Mg++ ions should exceed that of total dNTP concentration by 0.5 mM - 3.0 mM.The main source of phosphate groups in a reaction is the dNTPs so any change in their concentration affects the concentration of available Mg++ since Mg++ form a soluble complex with dNTPs. • Mg++ has been shown to be a superior divalent cation compared to Mn++ and Ca++ and will bind to the template DNA, dNTPs, primers, and polymerase.Its concentration affects product specificity, primer annealing, the formation of primer-dimer artifacts, melting temperature, and enzyme activity and fidelity. Thus, an important optimization strategy is to titrate your magnesium concentration for each DNA template, dNTP, primer and/or polymerase concentration change - especially if large products (>1 kb) are being amplified. • Improve specificity: Decrease concentration. Note that inadequate Mg++ concentration can result in lower efficiency/yields. • Improve efficiency: Increase concentration. However, excess Mg++ tends to cause nonspecific reactions and smeared electrophoretic bands, lowering specificity.
. DNTPs concentration : inbalanced dNTPs mixtures reduce Taq fidelity dNTPs reduce free Mg‡ interfering polymerase activity & decreasing primer annealing
Deoxynucleoside Triphosphates (dNTPs) • Standard Range: 20 -200 µM dNTP concentration should be titrated together with the primers. If one dNTP is at a higher concentration it will be preferentially incorporated for this reason, the four dNTP concentrations (dATP, dCTP, dTTP, dGTP) should be the same so that accurate incorporation takes place. In addition, it is important to keep the four dNTP concentrations above the estimated Km of each dNTP (10 µM - 15 µM). • Improve specificity: Reduce dNTP concentration but note that the Mg++ concentration should be lowered in an equimolar proportion. This is because the main source of phosphate groups in a reaction is the dNTPs so any change in their concentration affects the concentration of available Mg++ since Mg++ form a soluble complex with dNTPs. • Improve efficiency: For a large template sequence, increase the amount of dNTP. • Improve fidelity: Lower dNTP concentrations but note that the Mg++ concentration should be lowered in an equimolar proportion.
Annealing Temperature • Standard Range: 37°C - 65°C, 10 - 120 secs. During annealing, the primers are rapidly hybridized. Differences in the Km possessed by different DNA polymerases can change the effective primer annealing temperature. • Annealing times for LA PCR can be approximated with the formula: 60 secs + (2.5 secs/100 bases) = approximate annealing time • Improve specificity:Increase the annealing temperature (increments of 2°-5° C are recommended) since it reduces the possibilities of non-specific priming and therefore nonspecific product formation. Reduce annealing times since very long annealing times normally do not improve yield, but rather produce an increase in spurious priming and thus greater amounts of nonspecific PCR products.
Thermal cycling profile • Initial denaturation: completely denature complex genomic DNA accessible for primers • Primer annealing: the most critical factor in designing a high specificity PCR • Primer extension:- opt.temp for Taq DNA pol. ~ 72°C • - +60 bases/sec. 20” ~ <500 bp • . Denaturation step during cycling: • - Usually 95°C ~ 20”-30”, • too low incomplete snaps back no acces for primers
Final extension : • usually 72°C 5-15’ • Promote completion of partial extension products and annealing single stranded complementary products
Template: • conc.should optimized • Too little primers may not able to find target • Too much lead to increase mispriming • Template purity influences outcome of reaction. • General work strategies to avoid contamination • Laboratory bench & equipments • Cross contamination between samples • Product from previous PCR amplification
COMPONENT VOLUME FINAL CONCENTRATION 1.autoclaved ultra-filtered water (pH 7.0) 20.7µL - 2.10x PCR Buffer* 2.5µL 1x 3.dNTPs mix (25 mM each nucleotide) 0.2µL 200 µM (each nucleotide) 4.primer mix (25 pmoles/µL each primer) 0.4µL 0.4 µM (each primer) 5.Taq DNA polymerase (native enzyme) 0.2µL 1 Unit/25 µL 6.genomic DNA template (100 ng/µL) 1.0µL 100 ng/25 µL Example of a PCR Protocol * The 10x PCR buffer contains: 500 mM KCl; 100 mM Tris-HCl (pH 8.3); 15 mM MgCl2 (the final concentrations of these ingredients in the PCR mix are: 50 mM KCl; 10 mM Tris-HCl; 1.5 mM MgCl2).