Download

1 / 35
350 likes | 515 Views
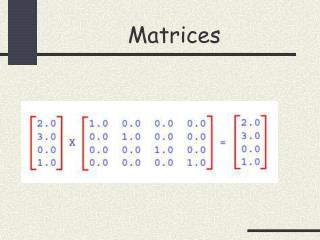
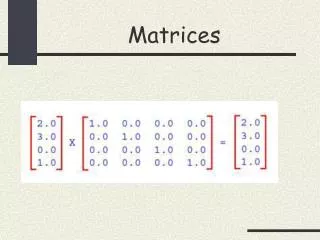
Dot Plots, Path Matrices, Score Matrices. Sequence A. V. T. R. I. V. H. V. N. S. I. L. P. S. T. N. I. L. S. V. I. L. S. T. R. I. Sequence B. V. I. L. P. E. F. S. T. diagonal lines give equivalent residues. Sequence A. V. T. R. I. V. H.

E N D
Dot Plots, Path Matrices, Score Matrices Sequence A V T R I V H V N S I L P S T N I L S V I L S T R I Sequence B V I L P E F S T diagonal lines give equivalent residues
Sequence A V T R I V H V N S I L P S T N I L S V I L S T R Sequence B I V I L P E F S T identical residues score 1 highest scoring path across the matrix gives best alignment
Sequence A V I L S L V I L P Q R S L V V I L S L V I L A L T V S T V I L S L V R N V I L P Q R I L S L V I S L A L Sequence B runs (tuples) of 3 residues 3 6 6 3 3 5 6 6 gap penalty = 3 SCORE = 20 - 9 = 11 3
Alignment from Dot Plot VILSLV ILPQRSLVVILSLVI LALTV STVILSLVNVILPQR ILSLVISLAL score = 20 sequence identity = 20/26 = 75%
ALVKRH… A H C N I R Q C L C R P M A 1 0 0 0 0 0 0 0 0 0 0 0 0 I 0 0 0 0 1 0 0 0 0 0 0 0 0 C 0 0 1 0 0 0 0 1 0 1 0 0 0 I 0 0 0 0 1 0 0 0 0 0 0 0 0 N 0 0 0 1 0 0 0 0 0 0 0 0 0 R 0 0 0 0 0 1 0 0 0 0 1 0 0 C 0 0 1 0 0 0 0 1 0 1 0 0 0 K 0 0 0 0 0 0 0 0 0 0 0 0 0 C 0 0 1 0 0 0 0 1 0 1 0 0 0 R 0 0 0 0 0 1 0 0 0 0 1 0 0 H 0 1 0 0 0 0 0 0 0 0 0 0 0 P 0 0 0 0 0 0 0 0 0 0 0 1 0 1 0 0 0 0…… 1 1 Path or Score Matrix 1 …HRKVLA 1 Residue substitution matrix
Needleman & Wunsch A H C N I R Q C L C R P M A 1 0 0 0 0 0 0 0 0 0 0 0 0 I 0 0 0 0 1 0 0 0 0 0 0 0 0 C 0 0 1 0 0 0 0 1 0 1 0 0 0 I 0 0 0 0 1 0 0 0 0 0 0 0 0 N 0 0 0 1 0 0 0 0 0 0 0 0 0 R 0 0 0 0 0 1 0 0 0 0 1 0 0 C 0 0 1 0 0 0 0 1 0 1 0 0 0 K 0 0 0 0 0 0 0 0 0 0 0 0 0 C 0 0 1 0 0 0 0 1 0 1 0 0 0 R 0 0 0 0 0 1 0 0 0 0 1 0 0 H 0 1 0 0 0 0 0 0 0 0 0 0 0 P 0 0 0 0 0 0 0 0 0 0 0 1 0
Needleman & Wunsch Algorithm • Accumulate the matrix by adding to each cell the highest score in the column or row to the right and below it • find the highest scoring path in the matrix by: • starting in the top left corner • moving down across the matrix from cell to cell • choosing the highest scoring cell at each move • the path can not go back on itself or cross the same row or column twice
Accumulating the Matrix • Add to the score in the cell the highest score from a cell in the row or column to right and below i,j i-1,j-1 i-n,j-1 i-1,j-m
Sequence A A H C N I R Q C L C R P M A 8 7 6 6 5 4 4 3 3 2 1 0 0 I 7 7 6 6 6 4 4 3 3 2 1 0 0 C 6 6 7 6 5 4 4 4 3 3 1 0 0 I 6 6 6 5 6 4 4 3 3 2 1 0 0 N 5 5 5 6 5 5 4 3 3 3 1 0 0 R 4 4 4 4 4 5 4 3 3 2 2 0 0 Sequence B C 3 3 4 3 3 3 3 4 3 3 1 0 0 K 3 3 3 3 3 3 3 3 3 2 1 0 0 C 2 2 3 2 2 2 2 3 2 3 1 0 0 R 2 1 1 1 1 2 1 1 1 1 2 0 0 H 1 2 1 1 1 1 1 1 1 1 1 0 0 P 0 0 0 0 0 0 0 0 0 0 0 1 0
Possible Moves in Finding a Path across the Matrix • start in the leftmost or topmost row • move to the highest scoring cell in row or column to right and below i,j i-1,j-1 i-n,j-1 i-1,j-m
Sequence A A H C N I R Q C L C R P M A 8 7 6 6 5 4 4 3 3 2 1 0 0 I 7 7 6 6 6 4 4 3 3 2 1 0 0 C 6 6 7 6 5 4 4 4 3 3 1 0 0 I 6 6 6 5 6 4 4 3 3 2 1 0 0 N 5 5 5 6 5 5 4 3 3 3 1 0 0 Sequence B R 4 4 4 4 4 5 4 3 3 2 2 0 0 C 3 3 4 3 3 3 3 4 3 3 1 0 0 K 3 3 3 3 3 3 3 3 3 2 1 0 0 C 2 2 3 2 2 2 2 3 2 3 1 0 0 R 2 1 1 1 1 2 1 1 1 1 2 0 0 H 1 2 1 1 1 1 1 1 1 1 1 0 0 P 0 0 0 0 0 0 0 0 0 0 0 1 0
Sequence A A H C N I R Q C L C R P M A 8 7 6 6 5 4 4 3 3 2 1 0 0 I 7 7 6 6 6 4 4 3 3 2 1 0 0 C 6 6 7 6 5 4 4 4 3 3 1 0 0 I 6 6 6 5 6 4 4 3 3 2 1 0 0 Sequence B N 5 5 5 6 5 5 4 3 3 3 1 0 0 R 4 4 4 4 4 5 4 3 3 2 2 0 0 C 3 3 4 3 3 3 3 4 3 3 1 0 0 K 3 3 3 3 3 3 3 3 3 2 1 0 0 C 2 2 3 2 2 2 2 3 2 3 1 0 0 R 2 1 1 1 1 2 1 1 1 1 2 0 0 H 1 2 1 1 1 1 1 1 1 1 1 0 0 P 0 0 0 0 0 0 0 0 0 0 0 1 0 A H C N I - R Q C L C R - P M A I C - I N R - C K C R H P M
Searching Sequence Databases Do fast scans using approximate methods e.g.BLAST or PSIBLAST Align proteins carefully using a dynamic programming methodNeedleman & Wunsch Smith & Waterman Scan against sequence profiles (or HMMs) in secondary databases e.g.Pfam, Gene3D, InterPro Align query sequence against family relatives using:ClustalW, Jalview, MUSCLE, MAFFT Can you inherit functional information?
Profile Based Sequence Search Methods • by comparing related sequences within a protein family can identify patterns of conserved residues • even the most distant members of the family should have these patterns of conserved residues • can make a profile which encapsulates these patterns and use it to detect more distantly related sequences • highly conserved positions usually correspond to the buried core or functional residues within the active site
Iterated Application of BLAST PSI-BLAST Altschul et al. (1997) • first constructs a multiple alignment of all the related sequences identified by BLAST • then estimates the residue frequencies at each position to construct a score matrix Position Specific Score Matrices (PSSM) also known as weight matrices or profiles
PSI-BLAST Altschul et al. (1997) UniProt Database query sequence aligns matched sequences and builds profile further iterations pull out more distant sequence relatives
PSI-BLAST Use the Multiple Alignment to Calculate Residue Frequencies the residue frequencies at each position are used to calculate the scores for aligning a query sequence against the pattern query relatives putative relative P1……... P5 P6…………... Pn…………... three times more powerful than BLAST!!
Position specific substitution matrix 10 10 20 70 90 . …HRVLA A 10 I C I Path matrix or score matrix N R 70 C K C R 70 H 90 P
Multiple Alignment • direct extensions of the standard DP approach for the alignment of 2 sequences are computationally impossible for more than 3 sequences • practical heuristic solutions are based on the idea that sequences are evolutionary related and can be aligned using an underlying phylogenetic tree this is known as progressive alignment
(1) Pairwise Alignment B 4 sequences A, B, C, D D A 6 pairwise comparisons then cluster analysis A B C C D (2) Multiple Alignment following the tree from 1 B Align most similar pair D gaps to optimise alignment A Align next most similar pair C A new gap to optimise alignment of BD with AC C B Align alignments - preserve gaps D
Multiple Alignment • start by aligning the most closely related pairs using DP and gradually align these groups together keeping the gaps that appear in earlier alignments fixed • alternatively can add sequences one at a time to a growing multiple alignment the heuristic approach is not guaranteed to find the optimum alignment - but it is soundly based, biologically
ClustalW Higgins, 1997 • since the choice of parameters used can have significant effect on the alignment for very distant sequences, ClustalW addresses this problem by: position specific gap opening and extension penalties using different amino acid substitution matrices - one for close relatives, one for distant More recent resources: MAFFT MUSCLE JALVIEW
ClustalW Gap penalties • where structure is known, one would want to increase the gap penalty within helices and strands and decrease it between them - forcing gaps to occur more frequently in loops • if no structure known, can use simple rules which depends on the residues occurring and the frequencies of gaps e.g. use lower gap penalties where gaps already occur
Searching Protein Family Databases Secondary databases (as opposed to primary sequence databases) group proteins into related families Families are usually represented by a sequence profile or sequence model (Hidden Markov Model HMM) derived from a multiple sequence alignment of the relatives
Di Ii B Mi E HMMs for Protein Domain Family Recognition • Pfam, SUPERFAMILY, Gene3D : • Hidden Markov Models (HMMs) • sequence is aligned using a probabilistic model of interconnecting match, delete or insert states • contains statistical information on observed and expected positional variation - “fingerprint of a protein family”
Pfam-A Pfam-B Other • Pfam-A • 10,340 curated families with annotation • Pfam-B • 224,303 families derived from ADDA • (50% clearly related to a Pfam-A) • UniProt coverage • 74% of sequences • 51% of residues • PDB coverage • 94% of sequences • 76% of residues
Pfam : SEED alignment representative members Profile-HMM HMMer-2.0 Search UniProt FULL alignment Manually curated Automatically made
Pfam classification Protein Protein fold, etc.
Protein Pfam classification Protein fold, etc. Family
Protein Pfam classification Clan Protein fold, etc. Family
![[MATRICES ]](https://cdn4.slideserve.com/144276/matrices-dt.jpg)