Phylogenetic Tree
Phylogenetic Tree. Phylogenetic Tree: What it is. Drawing evolutionary tree from characteristics of organisms or some measured distances between them Represented as a tree where nodes are the organisms/objects and arcs are the proximity between the respective nodes


Phylogenetic Tree
E N D
Presentation Transcript
Phylogenetic Tree: What it is • Drawing evolutionary tree from characteristics of organisms or some measured distances between them • Represented as a tree where nodes are the organisms/objects and arcs are the proximity between the respective nodes • Based on how close the organisms are
Phylogenetic Tree: Motivation • Pure curiosity: biological science • One species can be studied for a related one: • Drug test on monkeys for human • Rare species can be spared in a study • Drug design on evolution of micro-organism: aids/flu vaccine/drug design depends on how do they evolve • Tracking pathogen sources • Genesis, archeology,,,
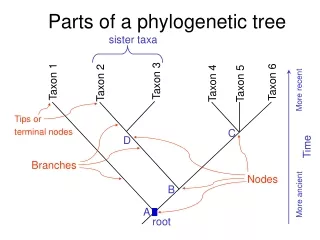
Phylogenetic Tree: topology • Evolutionary distance is not same as elapsed time: former is a crude approximation of the latter (if distance can be calculated at all) • Leaves are objects, internal nodes may or may not be objects (may represent hypothetical ancestors) • Mostly binary trees, sometimes not
Phylogenetic Tree: source data types • Discrete characters: • does it have long beaks? • Could be Boolean or multi-valued • Provided in matrix form (objects X characters) • Numerical distance matrix: • Symmetric pairwise distances measured by some means, e.g., by aligning sequences • Continuous character: character value is in numerical domain
Characters for phylogeny • Characters should be relevant in the context of phylogeny: depends on the user scientist • Characters should be independent: inherited without interference between the characters (eye color and hair color may not be a good combination in character set) • All characters must evolve from the same ancestor: we presume that (1) it is tree, (2) it is a connected tree • Closest objects are called “homologous”: max possible characters have same values or related values
Phylogeny using character state matrix • A “state” is a tuple with values for each character (value could be “unassigned”) • Internal node may be a state without any object assigned on it • Leaves are where the states correspond to objects with the respective assigned characters • P 178: a source character state matrix
Phylogeny using character state matrix: Problems • Convergence evolution: two non-homologous objects (most characters does not match, loosely speaking) happen to have same value on a character (needs a cycle in the graph)
Phylogeny using character state matrix: Problems • In one case evolution suggests character value of c evolves from “long” to “short,” in another case the reverse: confusion over the direction of evolution • Again, the tree property would be violated to accommodate this
Character domain types • Domain of character c could be: red < - > blue < - > yellow < - > green • C cannot evolve from blue to green without taking value yellow first • C is “ordered” • C can be directed and ordered, instead of undirected as above
Perfect phylogeny • Problem-free source • Each edge in phylogeny is a transition of the respective character’s value • All nodes with the same value for a character must form a subutree (with the transition at its root) • Such a tree is “perfect phylogeny”
Perfect phylogeny problem • Given a character state matrix does there exist a perfect phylogeny over it • P 178 table does not have a perfect phylogeny (presume transitions always 0 -> 1). Why? • P 180: table and its perfect phylogeny • What do you do when you do not have perfect phylogeny? Presume data is noisy and minimize errors in drawing perfect phylogeny
Perfect phylogeny problem • You can always try all possible trees over the objects and check whether each tree is perfect phylogeny or not • The total number of such trees is Pi[i=3 to n] (2i-5): Exponential
Perfect phylogeny problem: to check existence (Boolean matrix) • Organize char state matrix columnwise: for each col i set of objects is Oi • Every pair of Oi and Ok should be: • either Oi Ok • or Oi Ok • or Oi Ok = null • Either one belongs to another one or they do not overlap at all • If they overlap, no perfect phylogeny exist
Perfect phylogeny problem: to check existence (Boolean matrix) • In contrary, suppose Oi and Ok overlaps and a perfect phylogeny exists • say, i is the edge between (u, v): v and subtree has i=1, but all other nodes have i=0. • Suppose, three objects a, b, and c such that, a, b Oi, but c is not: a,b in subtree of v and c is not there • But, suppose b, c Ok, and a is not: b,c must belong to some other subtree separated by edge k • Contradiction
Perfect phylogeny problem: to check existence (Boolean matrix) • When no overlap exists: • Contained sets go within same subtree, if Oi Ok, then i-subtree is subtree of k-subtree • Disjoint sets are separate subtrees • Provesif and only if of the condition for perfect phylogeny • Algorithm for checking: Pairwise checking of object set may take O(m^2) for m characters, but set overlap may check even more time
Perfect phylogeny problem: Algorithm (Boolean matrix) • Sort the columns by number of 1’s (descending) • Scan each row to find which col number has the rightmost 1 for that box • Scan each column: every box should agree • Complexity O(mn) count, O(m log m) sort, O(mn) index matrix creation, O(mn) checking over index matrix: total O(mn) presuming n > log m
Perfect phylogeny problem: Algorithm (Boolean matrix) • Exercise: try the algorithm for tables 6.1 p 178 and 6.2 p 180 • Construction Algorithm: (1) sort characters/col increasing order, (2) each object – (3) each character – (4) if edge for char exists put obj on the end, (5) else create an edge and put object at the end, (6: cosmetic step) if more objects in a leaf node create edges for each object • O(nm) • Exc. Try it on table 6.2 p180
Perfect phylogeny problem: Algorithm (non-Boolean matrix, but…) • If two states per character but the order of transition not known, then presume an order: • majority state 0, minority 1 (more ancestors are available) • Same Lemma must be applied after this presumption: no overlapping set of objects
Phylogeny problem: arbitrary domain size, unordered characters • (Def) Triangulated graph: [no big hole] cycle with >3 vertices has a short-cut edge • Sub-trees of a tree form triangulated graph (as intersection graph?) • (Def) Intersection Graph over subsets: subsets are nodes and edges between pairs of overlapping subsets
Phylogeny problem: arbitrary domain size, unordered characters • Fig 6.7, p187 intersection graph for Table 6.3 p188 [not triangulated, yet] • (Def) c-Triangulated graph: Connect edges of intersection graph G where nodes are of different characters, and if the graph becomes now triangulated, then G is c-triangulated • Fig 6.7 is c-triangulated
Phylogeny problem: arbitrary domain size, unordered characters • Iff a character state matrix translates to a c-triangulated graph then it admits perfect phylogeny • Creating+checking c-triangulation is NP-hard (related to finding max-clique problem)
Phylogeny problem: arbitrary domain size, unordered characters: 2 characters • For 2 characters, the intersection graph is bi-partite • Perfect phylogeny means (iff) the state intersection graph is acyclic
Phylogeny construction: arbitrary domain size, unordered characters: 2 characters • Algorithm: • (1) Construct intersection graph • (2) make nodes for edges (intersection of the objects in old nodes now goes to the new nodes) • (3) connect new nodes if they have overlapping objects • (4) spanning tree of the graph is phylogeny • (5: cosmetic step) objects huddled on a node should be put on separate leaves • Try on Table 6.4 p190, and check against Fig 6.8 p189
When Perfect Phylogeny does not exist • Eliminate problematic characters: which ones, an optimization problem – min number of characters: Compatibility criterion • Minimize convergence (character goes back to its previous value): Parsimony criterion • Both NP-complete problems
When Perfect Phylogeny does not exist: Parsimony • Compatibility problem: Does there exist a subset of characters such that Lemma 6.1 (non-overlapping set of objects) is valid (or Perfect Phylogeny exists)? • Equivalent to K-clique problem: does there exist a connected-subgraph with K or more nodes?
When Perfect Phylogeny does not exist: Parsimony • Poly-transformation from Clique to compatibility problem: nodes to character, 3 objects for each edge with specific character values • Every pair of NP-complete problems have two way poly-trans • Compatibility can also be poly-trans to Clique: characters to nodes, non-overlapping (compatible) characters to edges
Phylogeny with Distance Matrix • Input is a distance matrix (square, symmetric) between all pair of objects, instead of character state matrix • Output is phylogeny with leaves as objects and arcs have distances as labels
Phylogeny with Distance Matrix • Additive matrix: when you can draw a tree where distance between every pair of leaves on the tree is the real distance on distance matrix • Matrices are unlikely to be additive in practice • For non-additive matrix, minimize deviation over the tree: NP-hard problem
Phylogeny with Distance Matrix • Typically we have 2 matrices: (1) upper bound on distances, and (2) for lower bounds • Metric space: • dij>0, dii=0, dij=dji, for all I, j • dij =< dik + dkj • Additive metric spaces follow 4 point condition: dij+dkl=dik+djl >= dil+djk
Phylogeny with Distance Matrix • Tree should have 3-degree internal nodes (Fig 6.9, p194) • Arc xy to be split proportionately at c, to add a node z by arc cz, so that distances xz, zy are proper
Phylogeny with Distance Matrix • Mxz = dxc + dzc • Myz = dyc + dzc • Mxy = dxc + dyc • Three equations, three unknowns dxc, dyc, dzc to be solved for • The tree drawn is unique for 3 objects x, y and z
Phylogeny with Distance Matrix • Adding 4th object w is same as adding 3rd object z: • Add between older objects x and y splitting xy at c2 • If c2 coincides with c, ignore this and redo the same between zc • Object w may hang (from c2) between xz or yz, but will not have 2 different opportunities
Phylogeny with Distance Matrix • The property of uniqueness of the tree remain valid for any k objects for k>4, for metric additive distance matrix • The algorithm may have to try all possible places to split an arc, but there will be a unique position, for metric additive space
Phylogeny: Ultrametric tree • Exc: Get MST of a complete graph over table 6.5 p195 • Ultrametric tree construction: • Input: Distance matrices for High cut-off Mh, Low cut-off Ml (table 6.6 p 201) • Output: Phylogeny where leaf-to-leaf distances are within the bounds provided by the 2 matrices (fig 6.16 p202)
Phylogeny: Ultrametric tree • Algorithm: • Compute MST T over Mh (algorithm?): provides basis for structure of the tree • Compute “cut-off” values between each edge on T using Ml: provides basis for distances on the tree edges • Compute the ultrametric tree U and find distance on each arc using the cut-offs
Phylogeny: Ultrametric tree • Step 2.1: input T, output is rooted tree R where internal nodes represent edges of T • Sort MST T by edge weights (from Mh) non-increasing • Pick up edges by the sort as root in each iteration • The path between the end nodes must go via the root: the two nodes edge should be in two different subtrees • Next edge in the sort to be picked up that has the corresponding node (x) on the respective side of the previous root (xy) • Until no edge for a node (x) is left (all such xy is picked up), then the node x is on a leaf
Phylogeny: Ultrametric tree • Step 2.2 (cut-off): • For each pair of nodes (x, y) look at the path in R • See which is the least common ancestor, say (ab) [note each internal node represents an edge] • Look up table Ml, if Ml_xy is more than current cut-off(ab) replace it with M_xy • In other words, the highest Ml value on any edge on the path from x to y in T should be its distance on the ultrametric tree • On example p201-202: root (ad) is updated for pairs of all nodes on the opposite sides EB(1), ED(1), AD(4), AB(3), CB(4), CD(3)
Phylogeny: Ultrametric tree • Step 3 (ultrametric tree): Recompute R again same way as before • But, now put distance on internal nodes • Height of an internal node is its cut-off / 2 • Note, computation of R starts with root downwards • Adjust distances between the nodes as heights are being calculated • Done
Comparing phylogenies • Two trees are expected to be isomorphic • All nodes should be on the leaves, if not make it so • Pick up a node u and its sibling v on T1 • Look for u in T2 and if its sibling is not v: return False • If the sibling is v then merge uv into its parent (an dremove subtree with u and v) • Continue bottom up until both T1 and T2 become single node trees, then return True