Download

1 / 44
460 likes | 1.15k Views
TRASTORNOS DE LA COAGULACION HEREDITARIOS. Dr. Oscar Diaz Cambronero Servicio de Anestesiología, Reanimación y Tratamiento del Dolor Sesión de Formación Continuada Valencia 27 de Junio del 2006. CLASIFICACION DE LOS TRASTORNOS DE LA COAGULACION. HEREDITARIOS : Hemofilia A Hemofilia B

E N D
TRASTORNOS DE LA COAGULACION HEREDITARIOS Dr. Oscar Diaz Cambronero Servicio de Anestesiología, Reanimación y Tratamiento del Dolor Sesión de Formación Continuada Valencia 27 de Junio del 2006 Sesión de Formación Continuada Valencia 27de Junio 2006
CLASIFICACION DE LOS TRASTORNOS DE LA COAGULACION HEREDITARIOS : • Hemofilia A • Hemofilia B • Enfermedad de Von Willebrand • Otros: deficiencia de otros factores afibrinogenemia Sesión de Formación Continuada Valencia 27de Junio 2006
CLASIFICACION DE LOS TRASTORNOS DE LA COAGULACION ADQUIRIDOS : • Coagulacion intravascular diseminada • Anticoagulacion perioperatoria • Coagulopatias intraoperatorias • Hemorragia por farmacos • Disfuncion plaquetaria • Purpura trombocitopenica • Deficiencia de vitamina K Sesión de Formación Continuada Valencia 27de Junio 2006
TRASTORNOS DE LA COAGULACION HEREDITARIOS • Suelen deberse a la disminucion o ausencia de un solo factor procoagulante. • Su manejo se ha de realizar en estrecha colaboracion con el hematologo. Sesión de Formación Continuada Valencia 27de Junio 2006
TRASTORNOS DE LA COAGULACION HEREDITARIOS • Generalmente el enfermo conoce que padece estas enfermedades, sobretodo en las formas graves. • No obstante las formas leves o moderadas pueden manifestarse durante la cirugia , de ahí la importancia de un diagnostico preoperatorio, para un adecuado manejo. Sesión de Formación Continuada Valencia 27de Junio 2006
TRASTORNOS DE LA COAGULACION HEREDITARIOS • La anamnesis y los antecedentes personales y familiares son basicos. • La exploracion fisica nos orienta sobre el origen del defecto: • Hemostasia primaria(vasos,plaquetas) petequias, telangiectasias, equimosis • Coagulación(factores) hematomas, hemartrosis Sesión de Formación Continuada Valencia 27de Junio 2006
TRASTORNOS DE LA COAGULACION HEREDITARIOS • ANALITICA: nos permite catalogar el defecto y determinar las concentraciones plasmaticas de factores deficitarios indispensables para un correcto tratamiento. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • Enfermedad recesiva ligada a X. • Afecta 2/10.000 recien nacidos varones. • Defecto genético heterogéneo a nivel clínico, determinado por la ausencia, deficiencia grave o mal funcionamiento del factor VIII. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • CLASIFICACION: según el nivel de factor VIII detectable en plasma. • Formas leves: 5-25% del valor normal. • Formas moderadas : 1- 4% del valor normal. • Formas graves : factor VIII no detectable en plasma. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • La clínica se caracteriza por hemorragias musculoesqueléticas y articulares tras traumatismos o cirugías. • La hemorragia hacia espacios cerrados puede cursar con compresión de nervios periféricos, vasos u obstrucción de la vía aérea. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • Diagnostico de sospecha ante una hemorragia inhabitual en pacientes varones. • Analítica: TTPA alargado con TP y recuento plaquetario normal. • Diagnostico definitivo con la determinación de factores de la coagulación. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • El tratamiento se realiza con concentrados de factor VIII, la cantidad necesaria depende del nivel plasmático previo. De ahí la importancia de titular los niveles de factores en el preoperatorio. • Dada la corta vida media ( aprox 8h) se suele recurrir a la perfusión continua. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • Crioprecipitados: poco usados por su menor bioseguridad, necesidad de grandes volúmenes • Concentrados de factor VIII: son la piedra angular del tto de la hemofilia, los de origen humano pueden transmitir hepatitis A y parvovirus B19, no así los de origen recombinante. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • Hay disponible factor VIII recombinante, sin riesgo de transmisión de enfermedades virales. • La desmopresina ( 0,3 µg/kg/ev) aumenta los niveles de factor VIII y VWF pudiendo ser útil en pacientes con hemofilia leve o moderada. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • Un 10-20% de los pacientes desarrollan anticuerpos frente al factor VIII tras la exposición al mismo.Se puede iniciar tto con altas dosis de concentrado de factor VIII, para saturar el Ac, otras opciones incluyen inmunosupresión con ciclofosfamida, plasmaféresis o inducción de inmunotolerancia. Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA A • Todas estas opciones necesitan tiempo, estando indicado en los casos muy productores de anticuerpos ( mas de 5 BU) la administración de factor VII recombinante activado (90-100 µg/ 2-3h) Sesión de Formación Continuada Valencia 27de Junio 2006
HEMOFILIA B • Trastorno ligado a X, patrón de herencia y clínica indistinguibles de la hemofilia A. • 1/30.000 recien nacidos varones. • Déficit de factor IX • TTPa alargado • Tto con concentrados de factor IX (vida media de 24h) o factor IX recombinante Sesión de Formación Continuada Valencia 27de Junio 2006
MANEJO ANESTESICO DE LAS HEMOFILIAS • Cuidadoso manejo y posicionamiento del paciente(IOT, vias venosas, sonda urinaria) • De elección anestesia general, aunque no se contraindica la anestesia locorregional. • Evitar la vía intramuscular, premedicación oral. Sesión de Formación Continuada Valencia 27de Junio 2006
MANEJO ANESTESICO DE LAS HEMOFILIAS • Adecuado estudio preoperatorio con valoración por hematólogo, titulación de factores, dosificación de factores según vida media, edad, tipo de cirugía… Sesión de Formación Continuada Valencia 27de Junio 2006
ENFERMEDAD DE VON WILLEBRAND • Se debe a la presencia de un factor Von Willebrand ( FvW ) defectuoso o en cantidad deficiente en el plasma. • Incidencia 2-3% de la población, siendo la coagulopatia congénita mas frecuente. Sesión de Formación Continuada Valencia 27de Junio 2006
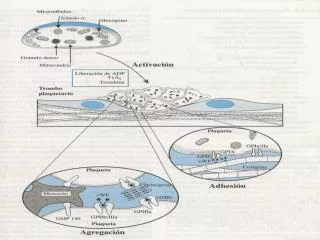
ENFERMEDAD DE VON WILLEBRAND • El FvW es un componente básico de la hemostasia primaria como enlace entre las plaquetas y el endotelio lesionado,se une al receptor GP Ib de la plaqueta y luego al GPIIb/IIIa del subendotelio expuesto. También participa en la hemostasia secundaria como transportador del factor VIII Sesión de Formación Continuada Valencia 27de Junio 2006
ENFERMEDAD DE VON WILLEBRAND • Clinicamente cursa con hemorragias mucosas, no siendo frecuentes los hematomas ni la hemartrosis. • Tiempo de hemorragia y TTPa alargado. • Existen varios tipos según el deficit, siendo necesario precisarlo para un correcto tto. Sesión de Formación Continuada Valencia 27de Junio 2006
ENFERMEDAD DE VON WILLEBRAND • Clasificación: • Tipo 1: déficit parcial cuantitativo, frecuencia del 70% ,reducción del FvW al 20-50% del valor normal, FvW funcionante. • Tipo 3: déficit absoluto cuantitativo, frecuencia 1-5%, es la forma mas grave asocia déficit severo de factor VIII Sesión de Formación Continuada Valencia 27de Junio 2006
ENFERMEDAD DE VON WILLEBRAND • Tipo 2: deficit cualitativo, 15-20%: • 2A : defecto cualitativo, • 2B : defecto cualitativo+trombocitopenia. • 2N o EvW Normandia: disminucion de la vida media del factor VIII de 12h a 1h, con disminucion de la union factor VIII FvW • 2M: defecto en la union FvW-GPIb • Tipo plaquetario o pseudo vWD: defecto a nivel del receptor GP Ib plaquetario Sesión de Formación Continuada Valencia 27de Junio 2006
ENFERMEDAD DE VON WILLEBRAND • DDAVP: la desmopresina (0,3µg/kg) es efectiva sobretodo en tipo1, estimula la liberación de FvW y factor VIII de las células endoteliales, aumentando la concentración plasmática 2 o 3 veces. Induce taquifilaxia al 4º-5º dia. Seria adecuado realizar un test de DDAVP previo a la cirugía para valorar el grado de respuesta. Sesión de Formación Continuada Valencia 27de Junio 2006
ENFERMEDAD DE VON WILLEBRAND • Concentrados de factor VIII: pacientes con mala respuesta a DDAVP como los tipos 2 y3, se benefician del uso de concentrados de factor VIII que contiene FvW, mejor que los crioprecipitados por el riesgo de infección viral. • Raramente en el tipo 3 se desarrollan Ac frente a Fvw, pudiendo usarse en estos casos factor rVIIa. Sesión de Formación Continuada Valencia 27de Junio 2006
AFIBRINOGENEMIA • Herencia autosómica recesiva • Heterocigotos: hipofibrinogenemia, • Homocigotos: Afibrinogenemia • Clinica: hemorragias graves( sangrado persistente del cordon umbilical) • T.hemorragia, TTPa,TP alargados • TTO: crioprecipitados o concentrados de fibrinógeno. Sesión de Formación Continuada Valencia 27de Junio 2006
DEFICIT DE PROTROMBINA • Herencia autosómica recesiva • Vitamina K dependiente • Clínica cutaneomucosa • TTPa y TP alargado con TT normal • Tto: plasma fresco congelado (riesgo de enfermedades virales), o complejo protrombínico ( riesgo de trombosis) Sesión de Formación Continuada Valencia 27de Junio 2006
DEFICIT DE FACTOR V (LEIDEN ) • Herencia autosómica recesiva • Clínica: hemorragias cutaneomucosas, postraumáticas, postquirúrgicas.. • T.hemorragia, TTPa, TP alargados • TTO: es la única coagulopatia que se trata con plasma fresco congelado por carecer del factor V Sesión de Formación Continuada Valencia 27de Junio 2006
DEFICIT DE FACTOR VII • Herencia autosómica recesiva • TP alargado co TTPa normal. • Clínica cutaneomucosa y menorragias • Tto con concentrados de complejo protrombínico, plasma fresco y en la actualidad con factor rVIIa (15-30 mg/kg cada 4-6 horas). Sesión de Formación Continuada Valencia 27de Junio 2006
DEFICIT DE FACTOR X • Herencia autosómica recesiva • Formas adquiridas en hepatopatias, amiloidosis, exposición a fungicidas.. • Clínica cutaneomucosa • TTPa y TP alargado • Tto: plasma fresco congelado (riesgo de enfermedades virales), o complejo protrombínico ( riesgo de trombosis) Sesión de Formación Continuada Valencia 27de Junio 2006
DEFICIT DE FACTOR XI • Herencia autosómica recesiva • Poca correlación entre la tasa de factor y las manifestaciones hemorrágicas. • Tto: fármacos antifibrinolíticos, plasma y en casos graves concentrados de factor XI Sesión de Formación Continuada Valencia 27de Junio 2006
DEFICIT DE FACTOR XII(Factor Hageman) • No se asocia con clínica hemorrágica pese al alargamiento del TTPa. • No necesitan tratamiento sustitutivo • Paradojicamente presentan riesgo de complicaciones trombóticas. Sesión de Formación Continuada Valencia 27de Junio 2006
DEFICIT DE FACTOR XIII • Es el factor estabilizador de la fibrina • Herencia autosómica recesiva, solo los homocigotos tienen clínica, sangrado por el cordón umbilical, hemorragias intracraneales espontaneas… • Pruebas de hemostasia habituales normales • Tto: crioprecipitado o concentrado especifico de factor XIII, vida media 19 días, realizar tto mensual Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) ¿ CUANDO ESTA INDICADO? Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) • Ficha técnica: • Hemofilia congénita con inhibidores a los factores VIII o IX > 5 UB • Hemofilia adquirida • Déficit de factor VII • Trombastenia de Glanzmann con Ac a GPIIb-IIIa y/o HLA y con historia previa o actual de resistencia a transfusiones de plaquetas. Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) • El factor rVIIa se cree que actua localmente en la misma lesión endotelial uniendose al factor tisular expuesto generando pequeñas cantidades de trombina que son suficientes para activar la superficie plaquetar produciendo mas trombina y asi activando la coagulación. Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) • El factor rVIIa se usa fuera de ficha técnica en pacientes sin anomalias previas de la coagulación pero que presentan un sangrado muy importante que llega a comprometer la vida y no se resuelve con medidas habituales. Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) • Se han descrito fuera de ficha técnica : • Enfermedad hepática (transplante hepático, biopsia hepática, HDA) • Cirugía y trauma severo • Reversión de terapia anticoagulante (fondaparinux, antagonistas vit K) • Sangrado masivo que compromete la vida Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) Dosis: 90µg/kg bolo Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) • Efectos secundarios: • La eficacia del Factor rVIIa en activar la coagulación nos obliga a pensar en las potenciales complicaciones tromboembólicas asociadas a su uso. • En pacientes hemofílicos aprox 1%. • En pacientes sin trastornos previos de la coagulación 1,4%. Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) • El factor rVIIa es un potente agente prohemostático que puede usarse en coagulopatias severas para detener o prevenir el sangrado, sobretodo en pacientes hemofílicos con inhibidores debiendo considerarse la primera linea de tratamiento. Sesión de Formación Continuada Valencia 27de Junio 2006
FACTOR VII RECOMBINANTE ACTIVADO (NOVOSEVEN®) • En pacientes sin déficits de la coagulación previos no esta claro el momento en que se ha de iniciar el tto, hasta que no haya mas evidencia disponible el uso de rVIIa fuera de ficha técnica se ha de restringir a situaciones con riesgo vital en las que el resto de opciones terapeuticas hayan fallado. Sesión de Formación Continuada Valencia 27de Junio 2006
GRACIAS POR SU ATENCION Sesión de Formación Continuada Valencia 27de Junio 2006