Download

1 / 7
80 likes | 227 Views
Denovo Sequencing Practical. Overview. V ery small dataset from Staphylococcus aureus 4 million x 75 base-pair, paired end reads Cover basic aspects of de-novo assembly from Illumina reads Does not cover Mixing other data types ( 454, Sanger, etc )

E N D
Overview • Very small dataset from Staphylococcus aureus • 4 million x 75 base-pair, paired end reads • Cover basic aspects of de-novo assembly from Illumina reads • Does not cover • Mixing other data types (454, Sanger, etc) • Gap filling techniques for “finishing” • Measuring the accuracy of assemblies • It’s really just an ‘introduction to VELVET’
Steps • Run files thru FastQC and examine ONLY the quality by read position graph and determine if the sequencing run was good ‘overall’ • Then run the sequences through Trimmomatic • Clip Illumina sequencing adapter • Allow clipping of leading and trailing ends • Use sliding window (size 4) trimming and a minimum length of 35 reads to be kept
Look at the resultant FASTQ files using ‘more’ or ‘less’ - notice the read length differences • Merge and ‘sort’ trimmed reads (velvet needs one file with pairs following each other) • shuffleSequences_fastq.pla.fastqb.fastqall.fastq
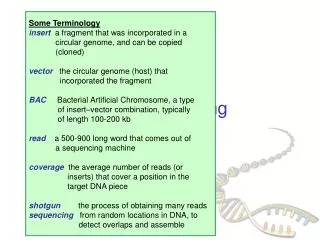
5. Run velveth • velveth auto 29,69,10-shortPaired–fastqall.fastq • Kmers of length 29 to 69 in increments of 10 • reads in these sequence file and simply produces a hashtableand • two output files • Roadmaps • Sequences • Needed by next program velvetg
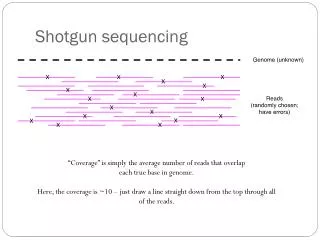
Run velvetg to determine best k of the various options • velvetgauto_<YOUR-KMER> -exp_cov auto -cov_cutoff auto • Example: • velvetg auto_39 -exp_cov auto -cov_cutoff auto • velvetg auto_69 -exp_cov auto -cov_cutoff auto • Runfasta_stats_N50.pl on the contigs • compare output logs between groups • Which k_mer length is the ‘best’? We will assume that the highest n50 reflects the optimal k_mer length In practice, we would use a finer granularity for the range tested
Bonus • Have a look at the velvet log and identify a long contig with highest coverage • Grab it in FASTA format and BLAST it against the nr protein database • What is the top hit? Is there any biological reason why it would have such high coverage?