Download
1 / 34
340 likes | 593 Views
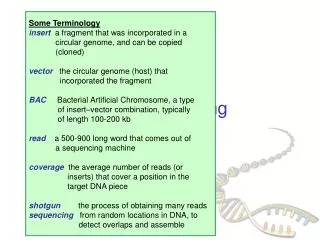
Some Terminology insert a fragment that was incorporated in a circular genome, and can be copied (cloned) vector the circular genome (host) that incorporated the fragment BAC Bacterial Artificial Chromosome, a type

E N D
Some Terminology insert a fragment that was incorporated in a circular genome, and can be copied (cloned) vector the circular genome (host) that incorporated the fragment BACBacterial Artificial Chromosome, a type of insert–vector combination, typically of length 100-200 kb read a 500-900 long word that comes out of a sequencing machine coveragethe average number of reads (or inserts) that cover a position in the target DNA piece shotgun the process of obtaining many reads sequencing from random locations in DNA, to detect overlaps and assemble DNA Sequencing
The Walking Method • Build a very redundant library of BACs with sequenced clone-ends (cheap to build) • Sequence some “seed” clones • “Walk” from seeds using clone-ends to pick library clones that extend left & right
Walking off a Single Seed • Cycle time to process one clone: 1-2 months • Grow clone • Prepare & Shear DNA • Prepare shotgun library & perform shotgun • Assemble in a computer • Close remaining gaps • A mammalian genome would need 15,000 walking steps !
Walking off several seeds in parallel • Few sequential steps • Additional redundant sequencing In general, can sequence a genome in ~5 walking steps, with <20% redundant sequencing Efficient Inefficient
Using Two Libraries Most inefficiency comes from closing a small gap with a much larger clone Solution: Use a second library of small clones
cut many times at random Whole Genome Shotgun Sequencing genome plasmids (2 – 10 Kbp) forward-reverse paired reads known dist cosmids (40 Kbp) ~500 bp ~500 bp
Advantages & Disadvantages of different sequencing strategies Physical Mapping • ADV. Easy assembly • DIS. Build physical map Whole Genome Shotgun (WGS) • ADV. No mapping • DIS. Difficult to assemble and resolve repeats Walking: combines some advantages of both Other possible method: • Shotgun sequencing of 10x BACs without any mapping • ADV. Can re-sequence hard regions • DIS. Too many shotgun libraries
Fragment Assembly Given N reads… Where N ~ 6 million… We need to use a linear-time algorithm
Steps to Assemble a Genome Some Terminology read a 500-900 long word that comes out of sequencer mate pair a pair of reads from two ends of the same insert fragment contig a contiguous sequence formed by several overlapping reads with no gaps supercontig an ordered and oriented set (scaffold) of contigs, usually by mate pairs consensus sequence derived from the sequene multiple alignment of reads in a contig 1. Find overlapping reads 2. Merge some “good” pairs of reads into longer contigs 3. Link contigs to form supercontigs 4. Derive consensus sequence ..ACGATTACAATAGGTT..
1. Find Overlapping Reads aaactgcagtacggatct aaactgcag aactgcagt … tacggatct gggcccaaactgcagtac gggcccaaa ggcccaaac … ctgcagtac gtacggatctactacaca tgacggatc gacggatct … tactacaca (word, read, orient., pos.) aaactgcag aactgcagt actgcagta … gtacggatc tacggatct gggcccaaa ggcccaaac gcccaaact … actgcagta ctgcagtac gtacggatc tacggatct acggatcta … ctactacac tactacaca (word, read, orient., pos.) aaactgcag aactgcagt acggatcta actgcagta actgcagta cccaaactg cggatctac ctactacac ctgcagtac ctgcagtac gcccaaact ggcccaaac gggcccaaa gtacggatc gtacggatc tacggatct tacggatct tactacaca
T GA TACA | || || TAGA TAGT 1. Find Overlapping Reads • Sort all k-mers in reads (k ~ 24) • Find pairs of reads sharing a k-mer • Extend to full alignment – throw away if not >97% similar TAGATTACACAGATTAC ||||||||||||||||| TAGATTACACAGATTAC
1. Find Overlapping Reads One caveat: repeats A k-mer that appears N times, initiates N2 comparisons ALU: 1,000,000 times Solution: Discard all k-mers that appear more than c Coverage, (c ~ 10)
1. Find Overlapping Reads Create local multiple alignments from the overlapping reads TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAG TTACACAGATTATTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAG TTACACAGATTATTGA TAGATTACACAGATTACTGA
1. Find Overlapping Reads (cont’d) • Correcterrors using multiple alignment C: 20 C: 20 C: 35 C: 35 C: 0 T: 30 C: 35 C: 35 TAGATTACACAGATTACTGA C: 40 C: 40 TAGATTACACAGATTACTGA TAG TTACACAGATTATTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA A: 15 A: 15 A: 25 A: 25 - A: 0 A: 40 A: 40 A: 25 A: 25 • Score alignments • Accept alignments with good scores
repeat region 2. Merge Reads into Contigs Merge reads up to potential repeat boundaries Unique Contig Overcollapsed Contig
2. Merge Reads into Contigs • Overlap graph: • Nodes: reads r1…..rn • Edges: overlaps (ri, rj, shift, orientation, score) Remove transitively inferrable overlaps
Repeats, errors, and contig lengths • Repeats shorter than read length are OK • Repeats with more base pair diffs than sequencing error rate are OK • To make the genome appear less repetitive, try to: • Increase read length • Decrease sequencing error rate Role of error correction: Discards ~90% of single-letter sequencing errors decreases error rate decreases effective repeat content increases contig length
repeat region 2. Merge Reads into Contigs • Ignore non-maximal reads • Merge only maximal reads into contigs
2. Merge Reads into Contigs sequencing error • Ignore “hanging” reads, when detecting repeat boundaries repeat boundary??? b a
2. Merge Reads into Contigs ????? Unambiguous • Insert non-maximal reads whenever unambiguous
3. Link Contigs into Supercontigs Normal density Too dense Overcollapsed Inconsistent links Overcollapsed?
3. Link Contigs into Supercontigs Find all links between unique contigs Connect contigs incrementally, if 2 links
3. Link Contigs into Supercontigs Fill gaps in supercontigs with paths of repeat contigs
4. Derive Consensus Sequence TAGATTACACAGATTACTGA TTGATGGCGTAA CTA Derive multiple alignment from pairwise read alignments TAGATTACACAGATTACTGACTTGATGGCGTAAACTA TAG TTACACAGATTATTGACTTCATGGCGTAA CTA TAGATTACACAGATTACTGACTTGATGGCGTAA CTA TAGATTACACAGATTACTGACTTGATGGGGTAA CTA TAGATTACACAGATTACTGACTTGATGGCGTAA CTA Derive each consensus base by weighted voting (Alternative: take maximum-quality letter)
Some Assemblers • PHRAP • Early assembler, widely used, good model of read errors • Overlap O(n2)->layout (no mate pairs)->consensus • Celera • First assembler to handle large genomes (fly, human, mouse) • overlap->layout->consensus • Arachne • Public assembler (mouse, several fungi) • overlap->layout->consensus • Phusion • overlap->clustering->PHRAP->assemblage->consensus • Euler • indexing->Euler graph->layout by picking paths->consensus
Quality of assemblies Celera’s assemblies of human and mouse
History of WGA 1997 • 1982: -virus, 48,502 bp • 1995: h-influenzae, 1 Mbp • 2000: fly, 100 Mbp • 2001 – present • human (3Gbp), mouse (2.5Gbp), rat*, chicken, dog, chimpanzee, several fungal genomes Let’s sequence the human genome with the shotgun strategy That is impossible, and a bad idea anyway Phil Green Gene Myers
Next few lectures More on alignments Large-scale global alignment – Comparing entire genomes Suffix trees, sparse dynamic programming MumMer, Avid, LAGAN, Shuffle-LAGAN Multiple alignment – Comparing proteins, many genomes Scoring, Multidimensional-DP, Center-Star, Progressive alignment CLUSTALW, TCOFFEE, MLAGAN Gene recognition Gene recognition on a single genome GENSCAN – A HMM for gene recognition Cross-species comparison-based gene recognition TWINSCAN – A HMM SLAM – A pair-HMM