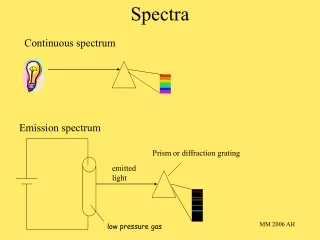
Vibrational Spectra
Vibrational Spectra. Molecules are not Static Vibration of bonds occurs in the liquid, solid and gaseous phase Vibrating Energy Frequency (and the appropriate frequencies for molecular vibrations are in the Infrared region of the electromagnetic spectrum


Vibrational Spectra
E N D
Presentation Transcript
Vibrational Spectra Molecules are not Static Vibration of bonds occurs in the liquid, solid and gaseous phase Vibrating Energy Frequency (and the appropriate frequencies for molecular vibrations are in the Infrared region of the electromagnetic spectrum Vibrations form therefore, a fundamental basis for spectroscopy in chemistry--the bonds are what makes the chemistry work in structure and function For Organic Chemistry the most important uses of these vibrations is for analysis of: •functional groups •structural identity, “fingerprinting” The IR spectra in this evenings talk are from the SDBS data base.
Tonight’s lecture Infrared Spectroscopy for Structure Determination A little theory Some notes on sampling Defer discussion of instrumentation Defer discussion of solids analysis Lots of examples, working through trends in related structure classes How to interpret, use data.
Some Perspective IR’s are not as thoroughly interpretable as NMR, Mass Spec Lacks the quantitative character on atom-atom basis that NMR has. (all the chromophores are not equal) Not used as much for identification as NMR and MS have become more accessible Still very useful for confirmation of structure cf. Reference spectra. Diagnostic for functional groups that may be silent or ambiguous in the NMR Quite sensitive, and can measure in all sorts of strange matrix e.g on surfaces, extremely useful for solid state characterization.
Infrared vs. Raman These two spectroscopies measure the same thing, vibrations, in different ways. IR is a absorption measurement, while Raman measures scattered light from a laser source, that in being scattered, is superimposed with the vibrational structure of the molecule. The selection rules are different--IR bands are active if in the act of the vibration, the dipole moment of the molecule changes. Raman band are active if the polarizability of the molecule changes. Often these two are complementary to each other Molecules of high symmetry frequently will not show IR activity
IR Measurement Neat film, or melt between two sodium chloride plates Solid solution in KBr, ground together and pressed into a transparent pellet Solution with appropriate blank region of solvent. Solution IR can be used to minimize broadening from self-association, H-bonding. Salt plates of CsCl2 for lower frequency window transparency (down to 200 cm-1) Mulling (grinding with mineral oil as dispersion), spread on salt plate Many different reflectance techniques, light must pass into the sample and reflect out.
Vibration levels are quantized, like everything else Like a harmonic oscillator With L-H, L moves most easily Can couple to other springs but heavy atoms can block or minimize this effect From Skoog and West
What Kind of vibrations are These? Bonds can……. These can number into the hundreds. Some are symmetrical, some antisymmetrical and many are coupled across the molecule Can be calculated. One easy approximation is: Bend Stretch k is the “force constant”, like the Hookes Law restoring force for a spring. Known and tabulated for different vibrations Wag (rock) The “reduced mass” where m1, m2 are the masses on either side of vibration
Regions of Frequencies Spectral Region Frequency(Hz) Wavenumber(cm-1) Wavelength (,m) After Table 16-1 of Skoog and West, et al. (Chapter 16)
What kinds of Bonds Absorb in which Regions? Bending is easier than stretching-- happens at lower energy (lower wavenumber) Bond Order is reflected in ordering-- triple>double>single (energy) with single bonds easier than double easier than triple Heavier atoms move slower than lighter ones The k in the frequency equation is in mDyne/Å of displacement Single bond str 3-6 mD/Å Double bond str. 10-12 mD/Å Triple Bond 15-18 mD/Å
The Fundamentals • These oscillating electric dipoles match in frequency the incoming e-field oscillations of IR light. • All the simple possibilities. For n atoms in a molecule; • Linear: 3n – 5 modes • Non-linear: 3n – 6 modes • Example for a methylene,given n=3 antisymmetric symmetric R R H H stretching R H R H R R H H in-plane bending R H R H scissoring rocking While useful, this oversimplifies, since molecular orbital picture requires that atoms can’t vibrate without affecting the rest of the molecule. R R H H out-of-plane R H R H bending wagging twisting
Calculated IR bands for CH2 in formaldehyde Formaldehyde spectrum from: http://www.cem.msu.edu/~reusch/VirtualText/Spectrpy/InfraRed/infrared.htm#ir2 Results generated using B3LYP//6-31G(d) in Gaussian 03W.
Looking at a Spectrum Divide the spectrum in to two regions: 4000 cm-1 1600 cm-1most of the stretching bands, specific functional groups (specific atom pairs). This is the “functional group” region. 1600 cm-1 400 cm-1Many band of mixed origin. Some prominent bands are reliable. This is the “fingerprint” region. Use for comparison with literature spectra. Wavenumber is cm-1=104/()
Nujol (mineral oil) CHCl3 CDCl3 Some IR Media are better than others Nujol, for example wipes out the hydrocarbon region for transparency.. Note that some older IR spectra while they are linear in frequency, the wavelength scale compresses the higher we go, Affects the appearance of spectra, not the line positions.
Reaction Monitoring Gas phase IR Pivaldehyde + methylamine Note loss of C=O Thank to Drs. Dalton and Mascavage
A Functional Group Chart 2000 800 group 4000 3600 3200 2800 1200 1600 2400 Cl I Br
The hydrogen stretching region Tip-- Draw a line straight up from 3000 cm-1. Intensity on left is Csp2-H, to the right is Csp3-H Arom- H 3000cm-1
Amines 3500, 3300 cm-1 doublet, frequently (without, with H-bonding effect) NH stretch 1600 cm-1 NH2 scissoring - broad 700-900 cm-1 NH2 wagging - broad, strong 1080 cm-1 C–N str. --weak for alkyl 1300 cm-1 Ar–N str. strong R-NH2 R–NH–R 3400 cm-1 singlet str. Weak C–N 1125 cm-1 No good IR bands, adj CH2 will shift to 2800 cm-1. A tert amine salt NH strong at 2500 cm-1 R–NR–R
Amine Salts R-NH3+ like methyl, but broad NH str. centered at 3000, br. deformation 1520-1570 br. R2NH2+ 3000 br, spikes at 2200,2500 cm-1 NH2 scissor at 1600 cm-1, broad
phenethanol 1-phenylethanol Alcohols C–O–H stretch 3600 cm-1 in dilute solution Typically H-bonding and at lower frequency ~3400 cm-1 C–O stretch in same region as C–C but much more intense Position is sensitive to subs. pattern RCH2–OH 1050 cm-1 R2CH–OH 1110 cm-1 R3C–OH 1175 cm-1 2-piperidinylethanol
Bands that should appear together SERVE AS CROSS CHECK e.g. see a triple bond? Check for C–H str. see C=O, check for OH, C–O I will point a few of these out as we go...
3,5-dimethyl-1-hexyn-3-ol 6-methyl5-hepten-1-yne phenylacetylene benzonitrile Phenyl-1-butyne Alkynes Also wk overtones at 1820,1790 cm-1
Functional Groups can be “NMR Silent” or ambiguous. IR can play a key role in Identification 1800, 1750 cm-1 (cyclic, has more intense 1750, acyclic, more instense 1800 C-O-C vs 1180-1220cm-1 1800 cm-1 doublet, Fermi resonance Poor resonance for 2p-3p, but strong inductive effect
More NMR silent groups--Nitro groups Analogous to Carboxylate ion. Strong bands Aromatic nitro 1520, 1350 cm-1 Aliphatic nitro 1550, 1370 cm-1
Double Bonds 1640 cm-1 is double bond stretch not seen for symmetrical molecules lower freq by conjugation, more intense =C–X lowers to 1590 cm-1 Ca. 890-910 cm-1 and 985cm-1 are o.o.p bendings for terminal =CH2 Cis vs Trans? Disubstituted –HC=CH- 960-970 cm-1 trans o.o.p bend 675-730 cm-1 cis o.o.p. bend Medium intensity
Methylenes 1460 cm-1 CH2 scissoring 725 cm-1 characteristic rocking for 4 or more CH2’s in a row (non-cyclic)
The Carbonyl Stretch is our friend… Carbonyl stretch changes its position for variation in specific structure THIS BAND IS ALWAYS STRONG!!! Good rules to remember… C=O conjugated to double bond goes lower in frequency With electronegative substituent (O, Cl) goes to higher frequency C=O in strained ring, goes to higher frequency C=O…(H hydrogen bonds lower the frequency)
O O O Ketones--sensitive to strain 1715 cm-1 1750 cm-1 1780 cm-1 Ca. 30 cm-1 higher for every C atom removed -diketones, str-str for open chain, IR inactive; in ring, 1720,1740 -haloketones--can see second band from rotamer populations (1720, 1745)
Ketones--Sensitive to conjugation 1660-1700 cm-1 rotational isomers cause doubling. S-trans 1674, S-cis 1699 1580-1640 cm-1 for enol 1715 cm-1 for the keto bond 1650-1700 cm-1 Along with br. OH str.
Ethyl-2-butenal cyclohexylcarboxaldehyde Aldehydes Doublet straddles 2800 cm-1 (Fermi resonance) Fundamental at ca. 2800 Bending at 1400, gives overtone at 2800
Hept-3-enoic acid Phenylbutyric acid Cyclohexanecarboxylic acid Carboxylic Acids 1715 cm-1 br OH stretch Good example of the broadening from H-bonding Also C—O 1280 cm-1, often a doublet O—H o.o.p bend br 920 cm-1 Salts have1600,1350 cm-1 broad!
Butyl acetate Esters and Lactones 1735 cm-1 1300-1100 intense, often doublet Cyclohexyl acetate
Effects of conjugation Lowers to 1715 cm-1 Raises to 1770 cm-1 Similar, to 1715 cm-1 : Weakens DB character Strengthens DB character (inductive over resonance)
Lactones, similar effects 1735 cm-1 1770 cm-1 1715 cm-1 1765 cm-1
N-benzylbenzamide phenylacetamide phenylacetanilide amides NH str 3300 cm-1 C=O 1650 cm-1 NH bend 1640 cm-1 Moves to 1550 for R-C(=O)-NHR’ L-a-aspartyl-L-phenylalanine 1-methyl ester
CH3 “umbrella” Ca. 1375 cm-1 “t-butyl split” “isopropyl split” isopropylcyclohexane t-butylcyclohexane Moves lower by 20 cm-1 methylcyclopentane Methyl Groups But… OCH3, NCH3 do not give this band
Complements the out-of plane bendings, related to the number of adjacent H… 750,700 750 5 750 From Crewes, Rodriguez and Jaspars, ch 8 4 750 850,780, 700 Out-of-plane bending combinations, quite small, but in a normally clean region of IR. Reliable even with nitro or carboxyl substitution 820 3 780 2 820 1 850 Unreliable with NO2, CO2H subs Benzene rings--substitution patterns Ring pucker at ca 700 cm-1is IR active for mono, 1,3-di-, 1,3,5-tri-, 1,2,3-tri- subs rings.
Putting the S in Silent; Sulfur containing groups wk SH at 2580 1360, 1180 cm-1 1050 cm-1 Extremely wk at 590-700 cm-1 1390, 1200 cm-1 1320, 1140 cm-1 Induction strengthens D.B. resonance not significant 1340, 1160 cm-1
Silence is Deafening(the IR guys think “silence is golden” R-N=C=O strong at 2250-2290 cm-1 R-N=C=S strong 2000-2190 cm-1 R-N=N=N strong 2100-2200 cm-1 R-N=C=N-R strong 2150 cm-1 R-CN 2250 cm-1 R-N+C:- 2130-80 cm-1 R-C=C=C-R’ (allenes) 1900-2000 cm-1 with very strong 850 wag if terminal