Download

1 / 39
390 likes | 402 Views
Explore how an organism's genome specifies behavior and characteristics through proteome analysis. Discover thousands of sequences, structural folds, functions, and expression patterns, all crucial for biological interactions and protein folding. Learn about de novo modeling, protein structure, and interaction predictions for insights into various biological systems.

E N D
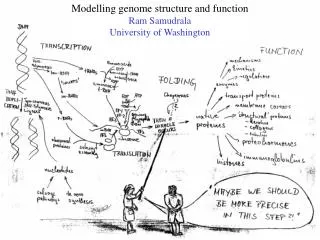
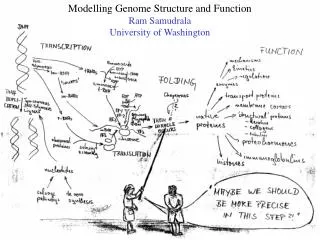
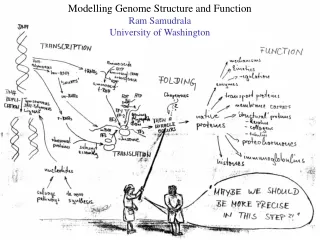
MODELLING PROTEOMES RAM SAMUDRALA ASSOCIATE PROFESSOR UNIVERSITY OF WASHINGTON How does the genome of an organism specify its behaviour and characteristics?
PROTEOME ~60,000 in human ~60,000 in rice ~4500 in bacteria Several thousand distinct sequence families
STRUCTURE A few thousand distinct structural folds
FUNCTION Tens of thousands of functions
EXPRESSION Different expression patterns based on time and location
INTERACTION Interaction and expression are interdependent with structure and function
PROTEIN FOLDING Protein sequence …-L-K-E-G-V-S-K-D-… One amino acid Unfolded protein Spontaneous self-organisation (~1 second) Native biologically relevant state Gene …-CTA-AAA-GAA-GGT-GTT-AGC-AAG-GTT-… • Not unique • Mobile • Inactive • Expanded • Irregular
PROTEIN FOLDING Spontaneous self-organisation (~1 second) Native biologically relevant state Gene …-CTA-AAA-GAA-GGT-GTT-AGC-AAG-GTT-… Protein sequence …-L-K-E-G-V-S-K-D-… One amino acid Unfolded protein • Not unique • Mobile • Inactive • Expanded • Irregular • Unique shape • Precisely ordered • Stable/functional • Globular/compact • Helices and sheets
STRUCTURE One distance constraint for every six residues One distance constraint for every ten residues 0 2 4 6 ACCURACY Experiment (X-ray, NMR) Computation (de novo) Computation (template-based) Hybrid (Iterative Bayesian interpretation of noisy NMR data with structure simulations) Cα RMSD
DE NOVO MODELLING Sample conformational space such that native-like conformations are found SELECT Hard to design functions that are not fooled by non-native conformations (“decoys”) Astronomically large number of conformations 5 states/100 residues = 5100 = 1070
DE NOVO MODELLING Make random moves to optimise what is observed in known structures GENERATE … … Find the most protein-like structures MINIMISE … … Filter based on all-atom pairwise interactions, bad contacts compactness, secondary structure, consensus of generated conformations FILTER
TEMPLATE-BASED MODELLING SCAN ALIGN KDHPFGFAVPTKNPDGTMNLMNWECAIP KDPPAGIGAPQDN----QNIMLWNAVIP ** * * * * * * * ** … … Build initial models from multiple templates using minimum perturbation Construct nonconserved side and main chains using graph theory and semfold Refine using constraints derived from multiple templates
STRUCTURE T0290 – peptidyl-prolyl isomerase from H. sapiens T0288 – PRKCA-binding from H. sapiens 2.2 Å Cα RMSD for 93 residues (25% identity) 0.5 Å Cα RMSD for 173 residues (60% identity) T0332 – methyltransferase from H. sapiens T0364 – hypothetical from P. putida 2.0 Å Cα RMSD for 159 residues (23% identity) 5.3 Å Cα RMSD for 153 residues (11% identity) Liu/Hong-Hung/Ngan
HYBRID MODELLING http://protinfo.compbio.washington.edu/protinfo_nmr http://protinfo.compbio.washington.edu/psicsi Hong-Hung
FUNCTION Ion binding energy prediction with a correlation of 0.7 Calcium ions predicted to < 0.05 Å RMSD in 130 cases Meta-functional signature accuracy Meta-functional signature for DXS model from M. tuberculosis Wang/Cheng
INTERACTION Transcription factor bound to DNA promoter regulog model from S. cerevisiae Prediction of binding energies of HIV protease mutants and inhibitors using docking with dynamics BtubA/BtubBinterolog model from P. dejongeii (35% identity to eukaryotic tubulins) McDermott/Wichadakul/Staley/Horst/Manocheewa/Jenwitheesuk/Bernard
SYSTEMS Example predicted protein interaction network from M. tuberculosis (107 proteins with 762 unique interactions) Proteins PPIs TRIs H. sapiens 26,741 17,652 828,807 1,045,622 S. cerevisiae 5,801 5,175 192,505 2,456 O.sativa (6) 125,568 19,810 338,783 439,990 E. coli 4,208 885 1,980 54,619 In sum, we can predict functions for more than 50% of a proteome, approximately ten million protein-protein and protein-DNA interactions with an expected accuracy of 50%. Utility in identifying function, essential proteins, and host pathogen interactions McDermott/Wichadakul
SYSTEMS Combining protein-protein and protein-DNA interaction networks to determine regulatory circuits McDermott/Wichadakul
INFRASTRUCTURE ~500,000 molecules over 50+proteomes served using a 1.2 TB PostgreSQL database and a sophisticated AJAX webapplication and XML-RPC API http://bioverse.compbio.washington.edu http://protinfo.compbio.washington.edu Guerquin/Frazier
INFRASTRUCTURE Guerquin/Frazier
INFRASTRUCTURE http://bioverse.compbio.washington.edu/integrator Chang/Rashid
APPLICATION: DRUG DISCOVERY CMV KHSV HSV Jenwitheesuk
APPLICATION: DRUG DISCOVERY Computionally predicted broad spectrum human herpesvirus protease inhibitors is effective in vitro against members from all three classes and is comparable or better than anti-herpes drugs CMV HSV KHSV Our protease inhibitor acts synergistically with acylovir (a nucleoside analogue that inhibits replication) and it is less likely to lead to resistant strains compared to acylovir HSV HSV Lagunoff
APPLICATION: NANOTECHNOLOGY Oren/Sarikaya/Tamerler
FUTURE + + Computational biology Structural genomics Functional genomics MODELLING PROTEIN AND PROTEOME STRUCTURE FUNCTION AT THE ATOMIC LEVEL IS NECESSARY TO UNDERSTAND THE RELATIONSHIPS BETWEEN SINGLE MOLECULES, SYSTEMS, PATHWAYS, CELLS, AND ORGANISMS
ACKNOWLEDGEMENTS Current group members: Past group members: Collaborators: • Baishali Chanda • Brady Bernard • Chuck Mader • David Nickle • Ersin Emre Oren • Ekachai Jenwitheesuk • Gong Cheng • Imran Rashid • Jeremy Horst • Ling-Hong Hung • Michal Guerquin • Rob Brasier • Rosalia Tungaraza • Shing-Chung Ngan • Siriphan Manocheewa • Somsak Phattarasukol • Stewart Moughon • Tianyun Liu • Vania Wang • Weerayuth Kittichotirat • Zach Frazier • Kristina Montgomery, Program Manager • Aaron Chang • Duncan Milburn • Jason McDermott • Kai Wang • Marissa LaMadrid • James Staley • Mehmet Sarikaya/Candan Tamerler • Michael Lagunoff • Roger Bumgarner • Wesley Van Voorhis Funding agencies: • National Institutes of Health • National Science Foundation • Searle Scholars Program • Puget Sound Partners in Global Health • UW Advanced Technology Initiative • Washington Research Foundation • UW TGIF
MOTIVATION FOR DETERMINING PROTEIN STRUCTURE The functions necessary for life are undertaken by proteins. Protein function is mediated by protein three-dimensional structure. Knowing protein structure at high resolution will enable us to: Determine and understand molecular function. Understand substrate and ligand binding. Devise intelligent mutagenesis and biochemical experiments to understand biological function. Design therapeutics rationally. Design novel proteins. Knowing the structures of all proteins encoded by an organism’s genome will enable us to understand complex pathways and systems, and ultimately organismal behaviour and evolution. Applications in the area of medicine, nanotechnology, and biological computing.
ALL-ATOM SCORING FUNCTION distance bins known structures atom-atom contacts AO AN AC … YOH 167 X167 contacts AO AN AC ... YOH AO AN AC … YOH s(dab) for contacts AO AN AC ... YOH candidate structure atom-atom contacts AO AN AC … YOH NxN contacts AO AN AC ... YOH
CRITICAL ASSESSMENT OF STRUCTURE PREDICTION Pre-CASP CASP Bias towards known structures Blind prediction
STRUCTURE TO FUNCTION? Hydrolase Ligase Lyase Oxidoreductase Transferase TIM barrel proteins 2000+ experimental structures
INTEROLOG MODELLING Interacting protein database Target proteome 85% Protein a Protein A Experimentally determined interaction Predicted interaction Protein b Protein B 90% Assign confidence based on similarity and strength of interaction Paradigm is the use of homology to transfer information across organisms; not limited to yeast, fly, and worm Consensus of interactions helps with confidence assignments
E. coli INTERACTIONS McDermott
M. tuberculosisINTERACTIONS McDermott
C. elegans INTERACTIONS McDermott
H. sapiens INTERACTIONS McDermott
Network-based annotation for C. elegans McDermott
KEY PROTEINS IN ANTHRAX Articulation points McDermott
HOST PATHOGEN INTERACTIONS McDermott