Download

1 / 18
180 likes | 201 Views
Explore methods for high-throughput protein structure determination using experimental and computational approaches for accurate results. Overcoming current limitations, such as insufficient protein quantities and complexities, by developing new computational and experimental methods. Utilize distance information from known structures, NMR, and mass spectrometry for efficient and automated structure determination in proteins.

E N D
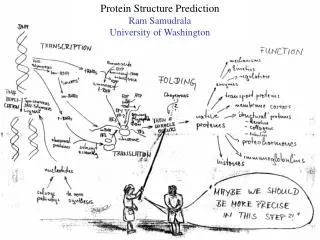
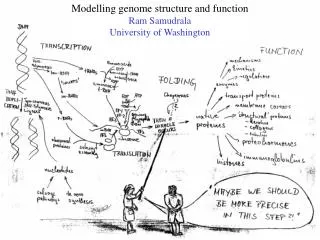
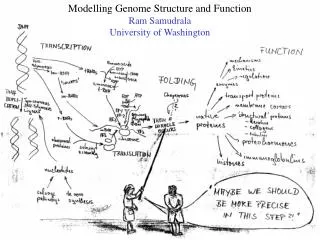
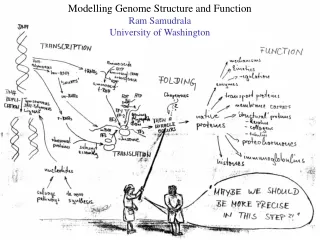
SHOTGUN STRUCTURAL PROTEOMICS RAM SAMUDRALA ASSOCIATE PROFESSOR UNIVERSITY OF WASHINGTON Given a heterogeneous mixture of proteins, how can we determine all their structures in a high-throughput and high-resolution manner?
METHODS FOR OBTAINING STRUCTURE One distance constraint for every six residues One distance constraint for every ten residues 0 2 4 6 ACCURACY Experiment (X-ray, NMR) Computation (de novo) Computation (template-based) Hybrid (Iterative Bayesian interpretation of noisy NMR data with structure simulations) Cα RMSD
WHY ARE CURRENT METHODS NOT ADEQUATE? The major bottlenecks for both X-ray diffraction and NMR studies is producing sufficient quantities of the protein in a pure form to perform the experiments. Deviations from ideal behaviour in a protein sample result in slow and labour-intensive structure determination, if at all possible. These major structure determination techniques were developed at a time when our worldview of proteins was simple and did not account for environment-dependent structure formation, protein dynamics and conformational changes, and post-translational modifications. The vast majority of proteins will therefore be inaccessible to X-ray diffraction and NMR studies. Computational approaches do not have the resolution of experimental approaches and lack consistency. Develop new methods based on crosslinking, mass spectroscopy, and isotope labelling for high throughput structure determination.
DISTANCE INFORMATION USING KNOWN STRUCTURES Residue specific all-atom probability discriminatory function (RAPDF) distance bins Known structures atom-atom contacts AO AN AC … YOH 167 X167 contacts AO AN AC ... YOH AO AN AC … YOH s(dab) for contacts AO AN AC ... YOH Candidate structure atom-atom contacts AO AN AC … YOH NxN contacts AO AN AC ... YOH
DISTANCE INFORMATION USING NMR Nucleii of proteins emit RF radiation measured in the form of chemical shifts. Primary source of distance information between protons is due to NOE. Steps: experiment (labourious), chemical shift assignment (automated), peak assignment (nontrivial), and structure determination (partially automated) . • HHN N • Peak coordinates: 1.2359.738130.97 • Protons with consistent chemical shifts: • 43 VAL HG1 1.256 - - • 8 ILE HN9.748 130.95 59 LEU HB3 1.242 - - Bayesian estimation of contact probabilities: Prior Post. Dist. 43 VAL HG1 - 8 ILE HN 0.038 0.75 4.6 Å 59 LEU HB3 - 8 ILE HN 0.002 0.05 8.0 Å
STRUCTURES USING COMPUTATION AND EXPERIMENT PROTINFO NMR structure for mjnop 3.5 Å Cα RMSD for 50 residues (required manual interpretation for several months) PROTINFO NMR structure for 1aye 1.8 Å Cα RMSD for 70 residues Bayesian approach calculates the probability distribution of each NOE peak contributing to proton-proton distances in a protein. Approach is assignment free, fast, fully automated, tolerant of noise, incompleteness and ambiguity, and enables iterative reinterpretation of source experimental data based on simulated structures (90% complete).
DISTANCE INFORMATION USING MASS SPECTROSCOPY MS MS Identify proteins with single crosslinks and fragment Identify crosslinked fragments Add crosslinkers VSKNT KEVN MS MKRS LVKQ Confirm sequence Repeat using different crosslinkers and isotope labelling
WHAT HAS BEEN DONE Proof of concept studies done by several people. A very good example: Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, and Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry.PNAS 97: 5802-5806, 2000. Eighteen intramolecular lysine-lysine crosslinks were identified for FGF-2 using crosslinking, MS, and proteolytic digestion, and fold identification. Authors claim method can be automated to produce structures in two days.
WHAT HAS BEEN DONE Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, and Dollinger G. PNAS 97: 5802-5806, 2000.
CROSSLINKING POSSIBILITIES Seven chemical groups that can be crosslinked from the following residues: cysteine, lysine, arginine, aspartate, glutamate, and the two terminii. Numerous distances for the 49 (7x7) possible pairs of groups. For every 100 residues, there may be up to ten members of each group, but typically only one crosslink is possible at a particular distance out of the ~100 possible pairs. A database of nonredundant protein structures reveals an average of 265 nonlocal crosslinks per protein and 1.5 per residue (estimate assuming a line of sight up to 20 Å between groups to be crosslinked).
HOW AND WHY WILL THIS WORK? Perform experiments to obtain a number of distance constraints for several proteins simultaneously. Perform simulations based on high confidence constraints and use distance distributions from resulting structures to iteratively reinterpret the spectra (without repeating experiment) until we obtain a high-resolution structure. Computational aspects largely complete. Components of approach have been implemented by others in a limited way but are assembled here in a robust and unique manner. Method can handle: Impure protein purification (ex: structural genomics failures). Environment-dependent structures (ex: chaperones + effectors). Partially disordered proteins. Several proteins simultaneously (large scale). No need for proteolytic digestion (complicates things). Focus on structures from noisy data, unlike X-ray diffraction and NMR.
OUR PROOF OF CONCEPT (IN PROGRESS) We have identified a novel herpesivirus protease inhibitor using docking with dynamics. We have experimentally verified this inhibitor works comparable to or better than existing antiherpes drugs against all representative members in cell culture. We have not verified whether inhibitor binds to active site of protease as predicted. We are synthesising, cloning, expressing, and purifying the protein (for Ki measurements). We will confirm presence or absence of bound inhibitor by crosslinking:
WHAT NEEDS TO BE DONE Crosslinkers need be constructed for several distances for all possible crosslinkable groups to get maximum number of constraints possible. Computational studies using simulated data (with noise) and develop software to prioritise experiments (ex: crosslinker choices). Initial studies starting with fairly pure mixtures >> not-so-pure mixtures >> 2-3 proteins >> handful of proteins >> Difficult proteins >> heterogenous mixtures >> whole proteomes. Bayesian framework utilised to estimate accuracy/error: Avoid repeating past oversight with NMR. Obtain an R-factor like estimate as in X-ray diffraction. Comparison of generated spectra from models to actual spectra. Iterative reinterpretation of experimental data.
OUTCOME AND EXPECTATIONS Structural genomics projects aim to obtain a representative structure of every protein family using X-ray diffraction and NMR methods and employ computational methods to fill in the gaps (enable coverage of the entire proteome). However, several families of proteins will not be accessible by these structure determination methodologies due to the need for large amounts of pure protein. Computational methods alone are far from capable of consistently producing high resolution structures. Even in successful cases, the dynamic effect of environmental effects on protein structure is not accounted for by current experimental and computational approaches. Our hybrid approach, which complements existing structural genomics efforts, will be used to rapidly obtain structures for entire proteomes in biologically relevant environments.
ACKNOWLEDGEMENTS Current group members: Past group members: Collaborators: • Baishali Chanda • Brady Bernard • Chuck Mader • David Nickle • Ersin Emre Oren • Ekachai Jenwitheesuk • Gong Cheng • Imran Rashid • Jeremy Horst • Ling-Hong Hung • Michal Guerquin • Rob Brasier • Rosalia Tungaraza • Shing-Chung Ngan • Siriphan Manocheewa • Somsak Phattarasukol • Stewart Moughon • Tianyun Liu • Vania Wang • Weerayuth Kittichotirat • Zach Frazier • Kristina Montgomery, Program Manager • Aaron Chang • Duncan Milburn • Jason McDermott • Kai Wang • Marissa LaMadrid • James Staley • Mehmet Sarikaya/Candan Tamerler • Michael Lagunoff • Roger Bumgarner • Wesley Van Voorhis Funding agencies: • National Institutes of Health • National Science Foundation • Searle Scholars Program • Puget Sound Partners in Global Health • UW Advanced Technology Initiative • Washington Research Foundation • UW TGIF
DISTANCE INFORMATION USING MASS SPECTROSCOPY Add labelled and unlabelled crosslinkers to a heterogeneous mixture of proteins Relative abundance mass/charge Repeat with different fragmentation resolution, crosslinker types, isotope labelling MS Enrich (LC, biotin) Relative abundance mass/charge For each peak representing a protein with a single crosslinker: fragment MS Identify peaks consistent with crosslinked fragments and obtain distance constraints
INTERPRETING MASS SPECTRA …AKRS…LKYVT…SKL…ARKT… (4 x 3 = 12 possibilities, one true contact) AKR-LK ARK-KL AKRS-LKY Relative abundance Relative abundance mass/charge mass/charge Ambiguous peaks in spectra are disambiguated (either eliminated or prioritised) using different fragmentation resolution, database preferences, and iterative reinterpretation after structure simulations AKR-LK ARK-KL AKR-LK ARK-KL AKR-SK? Relative abundance Relative abundance mass/charge mass/charge Spurious peaks in spectra are eliminated using isotope labelling (look for precise shifts)