Download

1 / 23
260 likes | 620 Views
Exome sequencing and complex disease :. practical aspects of rare variant association studies Alice Bouchoms Amaury Vanvinckenroye Maxime Legrand. What is exome sequencing ?. Exon : coding sequence of the DNA Exome sequencing :

E N D
Exomesequencing and complexdisease : practical aspects of rare variant association studies Alice Bouchoms Amaury Vanvinckenroye Maxime Legrand
Whatisexomesequencing ? • Exon : codingsequence of the DNA • Exomesequencing : • Aim : to sequence the coding part of the DNA i.e. the exons
Introduction • GWAS : helpeddiscovercommoncodingvariants • Exomesequencing • Also rare codingvariants • Faster, better • large sample ( > 10 000 individuals) • Before 2010 : only few publications on PUBMED • Now : more than2000 publications on PUBMED 2013 2012 2011
Study design • State objectives • Focus on extremeoutcomes • Unusualphenotype or traits • BUT : CAREFUL : de novo mutations • Geographical restrictions ?
Study design • Sequencingstrategy ? • Quality of the sample : 20x or greaterlevel of coverage depth of sequencing/person : 60x or greater • Non-codingregions : canstillbeusefull Determineancestries or estimategenotype • 0,2x to 2x
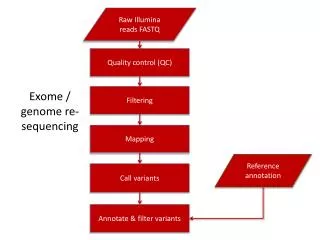
Variant calling • Goal : obtainhigh-qualitygenotypes • Severalsteps: • DNA contamination, DNA fingerprints, good follow-up? • Alignmentwithreferencegenome, calibration of base quality score, removal of duplicate reads.
Variant calling • Afterreadsmapping: • Samplequalitymetrics (spotting of outlierproperties) • Variant calling: • Look for differenceswhereoverlapsappear in alignmentwith the referencegenome
Variant calling • Machine-learning-based classifier: • Polymorphic variants / artifacts • Evaluate metrics : true / false positives • Quality metrics on samples • Recommendation: min depth of coverage 20X • Development of standards for storing sequence data and variant calls
Association analysis • Goal: find functional effects of variants • Score: indicates the effect on the protein function Separation between variants with high damage and the others • If multiple annotations, 3 ways: • Focus on the longest transcript • Focus on the most deleterious effect • Focus on the canonical transcript
Association analysis • Single variant association test Check of quality data • Usual way of processing rare variants: gather them in groups acting on the same gene to do the analysis
Association analysis • 2 methods for processing groups: • Comparison of the number of variantsbetween cases and controls • Comparisonwith chance expectations • Recommendation: at least a test of eachcategorywithdifferentthresholds • If no threshold, variety of frequencycut-offs
Association analysis • Packages available to perform the tests withsubsets of data • Example : • 1. missense, splice, stop alteringvariants • 2. subset of deleteriousvariants • 3. splice, stop alteringvariants
Association analysis • No optimal choices for the analysisbecause of variability of variants and of theircharateristicsbetweengenes. • Permutation-basedapproaches Statisticalsignificance • If no permutation-basedthreshold, p values ≤ 5 10-7 • QQ plots to summarize the results
Approaches for follow-up • To demonstrate association based on the analysedsamples, additionalsamples are needed.
Approaches for follow-up • Exome chip experiments examine most of the varaints, but not very sensitive to non-European populations.
Approaches for follow-up • Statistical imputation Take the base whichhas the highestcorrelationwith the missing one, and assume itis the sameallelethan T (i.e. minor or major). • But again, often not possible for mixed populations
Role of functionalassays • Study the changes in the proteins due to codingvariants • Studywhythese changes result in diverse diseases.
Forwardgenetics • Otherapproach to studyfunctionalvariants • First look atwhichproteins show changes • Thensearch in the DNA sequence for the variant(s)
Discussion • In other articles : • more careful about the samplequality • gain of sensitivity in variant calls if made amongseveralsamples • indels in variant call are the major source of false positive. Needalignmentalgorithmwhichallowsgapped alignement • Check results of association in data bases
Discussion • Because of costs, exomesequencingstudies focus on coding part of the genome. Thus not suitable for non-exonicsequence. (stucturalvariants, chromosomalrearrangements) • Theseproblemswillbepartiallysolvedby the cut in costs of sequencing