Download

1 / 12
120 likes | 190 Views
Learn how to predict protein structure using NMR chemical shifts through computational methods such as CamShift and molecular dynamics simulations. Discover the free energy surface and tools for accurate prediction in this tutorial.

E N D
Computational prediction of three-dimensional protein structure from NMR chemical shiftsKai KohlhoffMicrosoft Research Summer School CambridgeJuly 2008
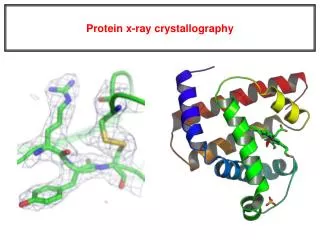
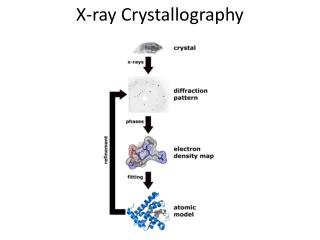
X-ray crystallography NMR spectroscopy
Molecular dynamics simulations Source: http://www.ch.embnet.org/MD_tutorial/
Free energy surface Source: http://www.lsbu.ac.uk/water/protein.html#fold
Donor n d CamShift chemical shift predictor
Energy Source: http://www.ch.embnet.org/MD_tutorial/ dexp - dcalc
MD CamShift-MD Energy Backbone RMSD (in Å = 0.1 nm) Truncated PVO (PDB 1PVO)
Acknowledgements Vendruscolo and Dobson group membersMichele VendruscoloAndrea CavalliPaul RobustelliXavier SalvatellaGian Gaetano TartagliaJoerg Gsponer Funding Microsoft Research