Download

1 / 20
210 likes | 576 Views
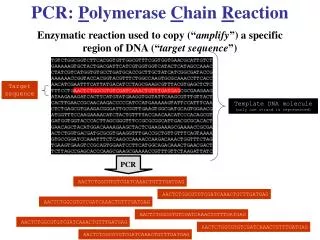
P olymerase C hain R eaction Enzyme that replicates DNA or RNA A reaction that, once started, uses its own product as a reactant… thus the reaction feeds itself leading to exponential growth How does PCR work? Start ( 1 copy ) 1. Melting (~94 °, 30 sec)

E N D
Polymerase Chain Reaction Enzyme that replicates DNA or RNA A reaction that, once started, uses its own product as a reactant… thus the reaction feeds itself leading to exponential growth
How does PCR work? Start (1 copy) 1. Melting (~94°, 30 sec) 2. Primer Annealing (~45°- 65°, 30 sec) 3. Primer Extension (~72°, 1 min per kb) Finish (2 copies)
Exponential amplification Start – 1 copy 4th Cycle – 16 copies 10th Cycle – 1024 copies 20th Cycle – 1.05 million copies 30th Cycle – 1.07 billion copies # of copies = 2n, (n = # of cycles) 1st Cycle – 2 copies 2nd Cycle – 4 copies 3rd Cycle – 8 copies
What is PCR used for? • The short answer: just about anything with DNA (or RNA) • Cloning into plasmids, etc. • cDNA creation • Mutagenesis • DNA (or RNA) quantification • Radioactive and non-radioactive labels • Screening bacterial clones for a correct plasmid • Genotyping • DNA sequencing
PCR Methods • Conventional PCR • Reverse-transcriptase (RT) PCR • Real-time PCR
Ingredients • ReactantsFinal concentration • PCR reaction buffer (10X) … dilute 1:10 • Magnesium (MgCl2 or MgSO4) ~1-5 mM (start with 2 mM) • dNTPs 0.2 mM each • Upstream primer ~0.5 mM • Downstream primer ~0.5 mM • Polymerase ~2.5 Units* • Template DNA 20-750 ng** • Mineral oil overlay? *follow instructions that come with polymerase **for plasmid DNA, use ~20 ng per 100uL reaction **for genomic DNA, use ~100-750 ng per 100uL reaction
Tweaking the reaction • Magnesium • Affects the binding of primers to the template DNA and the efficiency of the reaction • If you get little or no product, try repeating the reaction with a range of [Mg] • Template DNA • The purity of the template DNA will affect the reaction • Too much DNA can inhibit the reaction… sometimes less is more! • Primer Annealing temperature • Start out with this set a couple degrees lower than the melting temperature of your primers and adjust if necessary. • If you don’t get any product, try lowering the annealing temperature • If you get non-specific bands, try raising the annealing temperature
Polymerases Taq: old faithful, the original PCR polymerase now comes in every possible flavor (including Patton taq) High-fidelity: stays on the DNA better and has proofreading activity, so it amplifies longer templates with less errors (up to 12 kb) [examples: PWO, Taq HiFi, Pfu] “Hotstart”: polymerase is inactive until heated to 94°… this helps to reduce non-specific amplification (improvise this by not adding the polymerase until the first cycle has reached 94°)
Primers • How long do you need them to be? • 4n = chance that you will get a random match (n = length of primer) • 15 nt long = 1 match per ~1 billion base pairs • 20 nt long = 1 match per ~1 trillion base pairs (human genome is 3 billion) • Watch for primer homology and possible secondary structures • Primer pairs should not complement each other, especially at the 3’ end • Stretches of 5 G’s will form a secondary structure… also avoid repetitive sequences • Make sure you design the primers in the right orientation!!!
A/T = 10, G/C = 10 Tm = 55° A/T = 10, G/C = 10 Tm = 55° Designing primers ***Melting temperature shortcut: G or C = 4° A or T = 2° • Count the # of A/T and G/C in your primer • Tm = 2*(A+T) + 4*(G+C) - 5° • So, a 20 bp primer with 50% GC will have a Tm of 55° [2*10 +4*10 – 5 = 55] 5’-ACGATGCATGTAGCTACGTAGG…[YFG]…CGGGTACTGAACGGCAAACCTAGTC-3’ 3’-TGCTACGTACATCGATGCATCC….[YFG]…GCCCATGACTTGCCGTTTGGATCAG-5’
Controls • Negative Control (VERY IMPORTANT) • Add water instead of template DNA • Confirms that your reagents are not contaminated • Positive Control • If possible, run a parallel reaction with something that you know the primers will amplify
Experiment: • The DHFR gene is present in hamster CHOK-1 cells. You would like to put the hamster DHFR gene in human Hela cells and confirm integration by PCR. • You don’t think your primers will amplify a target from Hela cells, but you’d like to be sure. • You have the DHFR gene from CHOK-1 cloned onto a plasmid. DHFR (plasmid) CHOK-1 DNA Hela DNA Water (no DNA) Marker 1 Marker 2
Case 1 DHFR (plasmid) CHOK-1 DNA Hela DNA Water (no DNA) Marker 1 Marker 2 • Problem: No amplification • Positive control didn’t amplify, so reaction conditions are bad • Solutions? • Re-run reaction… maybe you forgot to add something • Lower primer annealing temperature • Check primer design • Try different Mg concentrations
Case 2 DHFR (plasmid) CHOK-1 DNA Hela DNA Water (no DNA) Marker 1 Marker 2 • Problem: Negative control has a band • Your reagents may be contaminated • Solutions? • Re-do the reaction… maybe you dropped a skin flake in the reaction mix or something • Discard old solutions and primers and use fresh materials • Set up the reaction away from where you usually do experiments… DNA around your work area can end up in your reactions
Case 3: screening clones Positive Control Clone #1 Clone #2 Clone #3 Clone #4 Clone #5 Water (no DNA) Marker • Problem: Too many bands • Your template DNA may be contaminated • Your primers may not be specific • Solutions? • Raise the annealing temperature to make your primers more specific • Try altering your primer design if possible • Try using a different sample of template DNA if possible
General tips • Set up your PCR reactions away from where you usually work • Use sterile technique and wear gloves • Filtered tips aren’t a bad idea • Design primers with similar melting temperatures • Add your template DNA last
PCR applications: radiolabelled probe • For Southerns, Northerns, etc. Probe sequence primers • Replace dATP with 32P dATP or use radiolabelled primers • Run PCR using normal conditions Radioactive probe complementary to your target sequence
PCR applications: DNA quantitation • Conventional PCR • Semi-quantitative at best… • You only measure the end product, which only gives you a rough idea of the amount of starting template • Real-time PCR • quantitative • Measures the production of dsDNA as it is made using a fluorescent dye, so you can monitor the reaction in “real-time” and accurately measure the amount of starting template
Core facilities • 740 Light Hall: get everything you need for PCR right here, including primers and kits • Real-time PCR thermocycler:
Got Questions? • Steven Gray • Ellen Fanning’s lab, 2325 Stevenson Center (on the far side of MRBIII, on the 3rd floor) • 343-5802 • Steven.j.gray@vanderbilt.edu