Download

1 / 43
450 likes | 1.02k Views
DNA, RNA, Protein Structure Prediction. Laura Pombo Laboratory of Computational Engineering Helsinki University of Technology . INTRODUCTION: Bioinformatics DNA RNA Proteins. BIOINFORMATICS.

E N D
DNA, RNA, Protein Structure Prediction Laura Pombo Laboratory of Computational Engineering Helsinki University of Technology
INTRODUCTION: Bioinformatics DNA RNA Proteins
BIOINFORMATICS • Bioinformatics involves the integration of computers, software tools, and databases in an effort to address biological questions. Bioinformatics approaches are often used for major initiatives that generate large data sets. • Two important large-scale activities that use bioinformatics are genomics and proteomics. • Genomics refers to the analysis of genomes. • A genome can be thought of as the complete set of DNA sequences that codes for the hereditary material that is passed on from generation to generation. • Thus, genomics refers to the sequencing and analysis of all of these genomic entities, including genes and transcripts, in an organism.
Bioinformatics, continue … • Proteomics, on the other hand, refers to the analysis of the complete set of proteins or proteome. • In addition to genomics and proteomics, there are many more areas of biology where bioinformatics is being applied (i.e., metabolomics, transcriptomics). Each of these important areas in bioinformatics aims to understand complex biological systems. Many scientists today refer to the next wave in bioinformatics as systems biology, an approach to tackle new and complex biological questions. Systems biology involves the integration of genomics, proteomics, and bioinformatics information to create a whole system view of a biological entity.
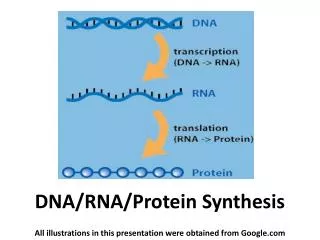
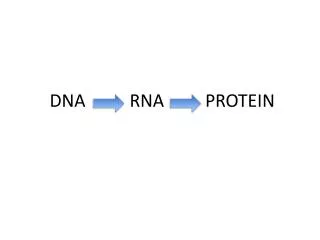
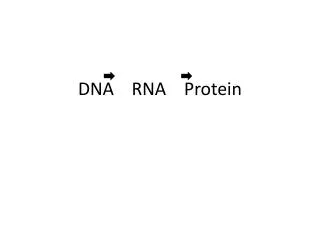
Central Dogma • DNA • RNA • Protein
DNA to RNA Portions of DNA Sequence Are Transcribed into RNA • The first step of a cell is to copy a particular portion of its DNA nucleotide sequence ( =gene) • Similarities: • DNA and RNA is a linear polymer made of four different types of nucleotide subunits linked together by phosphodiester bonds • DNA and RNA contains the bases adenine (A), guanine (G) and cytosine (C) • Differences: • In RNA the nucleotides are ribonucleotides (=contain the sugar ribose) • RNA contains uracil (U) instead of the thymine (T) My summary from the book: Molecular Biology of THE CELL (Bruce Alberts, et al.)
Different RNAs • mRNAs • (messenger RNAs), code for proteins • rRNAs • (ribosomal RNAs), form the basic structure of the ribosome and catalyze protein synthesis • tRNAs • (transfer RNA), central to protein synthesis as adaptors between mRNA and amino acids • snRNAs • (small nuclear RNAs), function in a variety of nuclear processes, including the splicing of pre-Mrna • snoRNAs • (small nucleolar RNAs), used to process and chemically modify rRNAs • Other noncoding RNAs • function in diverse cellular processes, including telomere synthesis, X-chromosome inactivation and the transport of proteins into te ER
http://gibk26.bse.kyutech.ac.jp/jouhou/image/dna-protein/all/N3utr.gifhttp://gibk26.bse.kyutech.ac.jp/jouhou/image/dna-protein/all/N3utr.gif
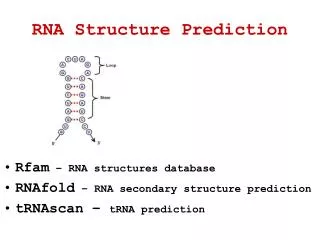
RNA is transcribed (or synthesized) in cells as single strands of (ribose) nucleic acids. However, these sequences are not simply long strands of nucleotides. Rather, intra-strand base pairing will produce structures. • In RNA, guanine and cytosine pair (GC) by forming a triple hydrogen bond, and adenine and uracil pair (AU) by a double hydrogen bond; additionally, guanine and uracil can form a single hydrogen bond base pair.
RNA structure prediction • Vienna RNA (PackageRNA Secondary Structure Prediction and Comparison) http://www.tbi.univie.ac.at/~ivo/RNA/ including a few precompiled binaries for download http://www.tbi.univie.ac.at/~ivo/RNA/windoze/ [under Windows] • The Vienna RNA Package consists of a C code library and several stand-alone programs for the prediction and comparison of RNA secondary structures. • RNA secondary structure prediction through energy minimization is the most used function in the package. • The program provides three kinds of dynamic programming algorithms for structure prediction: • the minimum free energy algorithm of (Zuker & Stiegler 1981) which yields a single optimal structure, • the partition function algorithm of (McCaskill 1990) which calculates base pair probabilities in the thermodynamic ensemble, and • the suboptimal folding algorithm of (Wuchty et.al 1999) which generates all suboptimal structures within a given energy range of the optimal energy.
RNAFOLD tool • RNAfold reads RNA sequences from stdin and calculates their minimum free energy (mfe) structure, partition function (pf) and base pairing probability matrix. It returns the mfe structure in bracket notation, its energy, the free energy of the thermodynamic ensemble and the frequency of the mfe structure in the ensemble to stdout. It also produces PostScript files with plots of the resulting secondary structure graph and a "dot plot" of the base pairing matrix. The dot plot shows a matrix of squares with area proportional to the pairing probability in the upper half, and one square for each pair in the minimum free energy structure in the lower half
ALIDOT program • Detecting Conserved RNA Structures The program alidot is designed to detect conserved RNA secondary structures in small data sets of related RNA sequences. The method is a combination of structure prediction and comparative sequence alignment. http://www.tbi.univie.ac.at/~ivo/RNA/ALIDOT/
http://images.google.com/imgres?imgurl=http://images.clinicaltools.com/images/gene/dna_versus_rna_reversed.jpg&imgrefurl=http://www.geneticsolutions.com/PageReq%3Fid%3D1530:1873&h=461&w=405&sz=135&tbnid=R7LVIZO4g6cJ:&tbnh=125&tbnw=109&hl=en&start=2&prev=/images%3Fq%3DDNA%2Bto%2BRNA%26svnum%3D10%26hl%3Den%26lr%3D%26sa%3DGhttp://images.google.com/imgres?imgurl=http://images.clinicaltools.com/images/gene/dna_versus_rna_reversed.jpg&imgrefurl=http://www.geneticsolutions.com/PageReq%3Fid%3D1530:1873&h=461&w=405&sz=135&tbnid=R7LVIZO4g6cJ:&tbnh=125&tbnw=109&hl=en&start=2&prev=/images%3Fq%3DDNA%2Bto%2BRNA%26svnum%3D10%26hl%3Den%26lr%3D%26sa%3DG
MEME • MEME is a tool for discovering motifs in a group of related DNA or protein sequences. A motif is a sequence pattern that occurs repeatedly in a group of related protein or DNA sequences. • MEME represents motifs as position-dependent letter-probability matrices which describe the probability of each possible letter at each position in the pattern. • Individual MEME motifs do not contain gaps. Patterns with variable-length gaps are split by MEME into two or more separate motifs. MEME takes asinputa group of DNA or protein sequences (the training set) and outputs as many motifs as requested. • MEME uses statistical modeling techniques to automatically choose the best width, number of occurrences, and description for each motif.
DNA structure prediction Other similar programs: Cassandra http://www-hto.usc.edu/software/procrustes/cassandra/cass_frm.html DNA Sequence Translation GENEID which predicts Gene Structure in Query Sequences (US) GRAIL, GenHunt, Censor, Pythia, Entrez, Beauty, etc. You should have a look in: http://restools.sdsc.edu/biotools/biotools16.html
PROTEIN • Protein: A large molecule composed of one or more chains of amino acids in a specific order determined by the base sequence of nucleotides in the DNA coding for the protein. • Proteins are required for the structure, function, and regulation of the body's cells, tissues, and organs. Each protein has unique functions. Proteins are essential components of muscles, skin, bones and the body as a whole. • Protein is one of the three types of nutrients used as energy sources by the body, the other two being carbohydrate and fat. Proteins and carbohydrates each provide 4 calories of energy per gram, while fats produce 9 calories per gram. • The word "protein" was introduced into science by the great Swedish physician and chemist Jöns Jacob Berzelius (1779-1848) who also determined the atomic and molecular weights of thousands of substances, discovered several elements including selenium, first isolated silicon and titanium, and created the present system of writing chemical symbols and reactions.
Tools for PROTEIN Structure Prediction: ExPASy • The ExPASy (Expert Protein Analysis System) proteomics server from the Swiss Institute of Bioinformatics (SIB) is dedicated to molecular biology with an emphasis on data relevant to proteins.It allows you to browse through a number of databases produced in Geneva, such as Swiss-Prot, PROSITE, SWISS-2DPAGE, SWISS-3DIMAGE, ENZYME, as well as other cross-referenced databases (such as EMBL/GenBank/DDBJ, OMIM, Medline, FlyBase, ProDom, SGD, SubtiList, etc). • It also allows access to many analytical tools for the identification of proteins, the analysis of their sequence and the prediction of their tertiary structure. ExPASy also offers you many documents relevant to these field of research and you will find from the servers, links to most relevant sources of information across the Web.Swiss-2DService is a non-profit 2-D PAGE service to the scientific community • ExPASy was created in August 1993, it was one of the first WWW servers for biological sciences. Since that date it has undergone constant modifications and improvements.
PROSITE Database of protein families and domains • PROSITE is a database of protein families and domains. • It consists of biologically significant sites, patterns and profiles that help to reliably identify to which known protein family (if any) a new sequence belongs. • It is based on the observation that, while there is a huge number of different proteins, most of them can be grouped, on the basis of similarities in their sequences, into a limited number of families. • Proteins or protein domains belonging to a particular family generally share functional attributes and are derived from a common ancestor.It is apparent, when studying protein sequence families, that some regions have been better conserved than others during evolution. http://au.expasy.org/prosite/prosite_details.html
These regions are generally important for the function of a protein and/or for the maintenance of its three- dimensional structure. By analyzing the constant and variable properties of such groups of similar sequences, it is possible to derive a signature for a protein family or domain, which distinguishes its members from all other unrelated proteins. • A pertinent analogy is the use of fingerprints by the police for identification purposes. A fingerprint is generally sufficient to identify a given individual. Similarly, a protein signature can be used to assign a newly sequenced protein to a specific family of proteins and thus to formulate hypotheses about its function. • PROSITE currently contains patterns and profiles specific for more than a thousand protein families or domains. Each of these signatures comes with documentation providing background information on the structure and function of these proteins.
Experimental Data • Disulphide bonds • Spectroscopic data • Site directed mutagenesis studies • Knowledge of proteolytic cleavage sites • Etc.
Protein sequence data • Is your protein a transmembrane protein, or does it contain transmembrane segments? Methods for predicting these segments: • TMAP (EMBL) • PredictProtein (EMBL/Columbia) • TMHMM (CBS, Denmark) • TMpred (Baylor College) • DAS (Stockholm) • Does your protein contain coiled-coils? Methods: • At COILS server, the COILS program • Does your protein contain regions of low complexity? Methods: • the program SEG
Sequence database searching 1/2 • Comparisons with sequence databases to find homologues, methods: • the BLAST suite of programs. • National Center for Biotechnology Information (USA) Searches • European Bioinformatics Institute (UK) Searches • BLAST search through SBASE (domain database; ICGEB, Trieste) • Other methods for comparing a single sequence to a database include: • The FASTA suite (William Pearson, University of Virginia, USA) • SCANPS (Geoff Barton, European Bioinformatics Institute, UK) • BLITZ (Compugen's fast Smith Waterman search) • Multiple sequence information • building a profile from some kind of multiple sequence alignment. Methods: • PSI-BLAST (NCBI, Washington) • ProfileScan Server (ISREC, Geneva) • HMMER Hidden Markov Model searching (Sean Eddy, Washington University) • Wise package (Ewan Birney, Sanger Centre; this is for protein versus DNA comparisons)
Sequence database searching 2/2 • Incorporating multiple sequence information – a MOTIF. • describes the key residues that are conserved and define the family. • Sometimes this is called a "signature". For example, "H-[FW]-x-[LIVM]-x-G-x(5)-[LV]-H-x(3)-[DE]" describes a family of DNA binding proteins. Methods: • PROSITE, ExPASy • EBI • Pre-prepared protein alignments, databases: • SMART (Oxford/EMBL) • PFAM (Sanger Centre/Wash-U/Karolinska Intitutet) • COGS (NCBI) • PRINTS (UCL/Manchester) • BLOCKS (Fred Hutchinson Cancer Research Centre, Seatle) • SBASE (ICGEB, Trieste)
Multiple Sequence Alignment • Some methods and tools: • EBI (UK) Clustalw Server • IBCP (France) Multalin Server • IBCP (France) Clustalw Server • IBCP (France) Combined Multalin/Clustalw • MSA (USA) Server • BCM Multiple Sequence Alignment ClustalW Sever (USA) • Alignments can provide: • Information as to protein domain structure • The location of residues likely to be involved in protein function • Information of residues likely to be buried in the protein core or exposed to solvent • More information than a single sequence for applications like homology modelling and secondary structure prediction.
Secondary Structure Prediction methods and links • Methods and tools: • PSI-pred (PSI-BLAST profiles used for prediction; David Jones, Warwick) • JPRED Consensus prediction (includes many of the methods given below; Cuff & Barton, EBI) • DSC King & Sternberg (this server) • PREDATORFrischman & Argos (EMBL) • PHD home page Rost & Sander, EMBL, Germany • ZPRED server Zvelebil et al., Ludwig, U.K. • nnPredict Cohen et al., UCSF, USA. • BMERC PSA Server Boston University, USA • SSP (Nearest-neighbor) Solovyev and Salamov, Baylor College, USA. • If no homologue of known structure from which to make a 3D model • to predict secondary structure to provide the location of alpha helices, and beta strands within a protein or protein family.
Fold recognition methods and links 1/2 • Methods: • 3D-pssm (this server) • TOPITS (EMBL) • UCLA-DOE Structre Prediction Server (UCLA) • 123D • UCSC HMM (UCSC) • FAS (Burnham Institute) • THREADER(Warwick) • ProFIT CAME (Salzburg) • Even with no homologue of known 3D structure, it may be possible to find a suitable fold for your protein among known 3D structures by way of fold recognition methods. Prediction of protein 3D structures is not possible at present, • and a general solution to the protein folding problem is not likely to be found in the near future. • However, it has long been recognised that proteins often adopt similar folds despite no significant sequence or functional similarity • There are numerous protein structure classifications now available via the WWW: • SCOP (MRC Cambridge) • CATH (University College, London) • FSSP (EBI, Cambridge) • 3 Dee (EBI, Cambridge) • HOMSTRAD (Biochemistry, Cambridge) • VAST (NCBI, USA) • The goal of fold recognition • Methods of protein fold recognition attempt to detect similarities between protein 3D structure that are not accompanied by any significant sequence similarity. There are many approaches, but the unifying theme is to try and find folds that are compatable with a particular sequence. Unlike sequence-only comparison, these methods take advantage of the extra information made available by 3D structure information. In effect, the turn the protein folding problem on it's head: rather than predicting how a sequence will fold, they predict how well a fold will fit a sequence. • The alignments that are output by the programs. They can be used as a starting point, but the best alignment of sequence on to tertiary structure is still likely to come from careful human intervention.
Fold recognition methods and links 2/2 • The goal of fold recognition • detect similarities between protein 3D structure that are not accompanied by any significant sequence similarity. • to find folds that are compatible with a particular sequence. • 3D structure information to predict how well a fold will fit a sequence. • the best alignment of sequence on to tertiary structure is still likely to come from human intervention.
Analysis of protein folds and alignment of secondary structure elements • to which fold your protein belongs, methods: • SCOP (MRC Cambridge) • CATH (University College, London) • FSSP (EBI, Cambridge) • 3 Dee (EBI, Cambridge) • HOMSTRAD (Biochemistry, Cambridge) • VAST (NCBI, USA) • If there is any functional similarity between your protein and any members of the fold, then you may be able to back up your prediction of fold
Alignment of sequence to tertiary structure • Starting with the alignment from the fold recognition method, and considering the alignment of secondary structures. • Proteins having similar three-dimensional structures with little or no sequence similarity can differ substantial with respect to the finer details of their structures (i.e. loops, precise orientation of side chains, orientation of secondary structures, etc.).
Comparative or Homology Modelling • If significant homology to another protein of known three-dimensional structure, • model of your protein 3D structure can be obtained via homology modelling. • To build models, if you have found a suitable fold via fold recognition • to generate models automatically using the very useful SWISSMODEL server; • WHAT IF (G. Vriend, EMBL, Heidelberg) • MODELLER (A. Sali, Rockefeller University) • MODELLER Mirror FTP site • Once you have a three-dimensional model, it is useful to look at protein 3D structures: methods: • GRASP Anthony Nicholls, Columbia, USA. • MolMol Reto Koradi, ETH, Zurrich, C.H. • Prepi Suhail Islam, ICRF, U.K. • RasMol Roger Sayle, Glaxo, U.K.