Download

1 / 116
1.28k likes | 1.97k Views
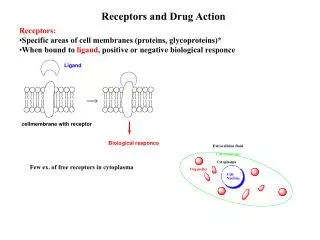
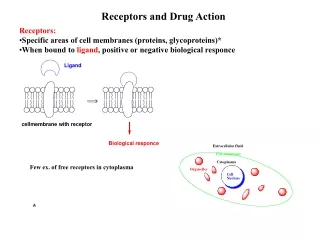
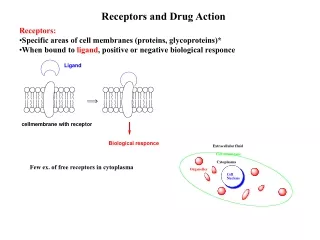
Chapter 8 Prodrugs and Drug Delivery Systems. The Organic Chemistry of Drug Design and Drug Action. Prodrugs and Drug Delivery Systems. Prodrug - a pharmacologically inactive compound that is converted to an active drug by a metabolic biotransformation

E N D
Chapter 8 Prodrugs and Drug Delivery Systems The Organic Chemistry of Drug Design and Drug Action
Prodrugs and Drug Delivery Systems Prodrug - a pharmacologically inactive compound that is converted to an active drug by a metabolic biotransformation Ideally, conversion occurs as soon as the desired goal for designing the prodrug is achieved. • Prodrugs and soft drugs are opposite: • a prodrug is inactive - requires metabolism to give active form • a soft drug is active - uses metabolism to promote excretion • A pro-soft drug would require metabolism to convert it to a soft drug
Utility of Prodrugs 1. Aqueous Solubility - to increase water solubility so it can be injected in a small volume 2. Absorption and Distribution - to increase lipid solubility to penetrate membranes for better absorption and ability to reach site of action 3. Site Specificity - to target a particular organ or tissue if a high concentration of certain enzymes is at a particular site or append something that directs the drug to a particular site
Utility of Prodrugs (cont’d) 4. Instability - to prevent rapid metabolism; avoid first-pass effect 5. Prolonged Release - to attain a slow, steady release of the drug 6. Toxicity - to make less toxic until it reaches the site of action 7. Poor Patient Acceptability - to remove an unpleasant taste or odor; gastric irritation 8. Formulation Problems - to convert a drug that is a gas or volatile liquid into a solid
Types of ProdrugsDrug Latentiation - rational prodrug design I. Carrier-linked prodrug A compound that contains an active drug linked to a carrier group that is removed enzymatically A. bipartate - comprised of one carrier attached to drug B. tripartate - carrier connected to a linker that is connected to drug C. mutual - two, usually synergistic, drugs attached to each other II. Bioprecursor prodrug A compound metabolized by molecular modification into a new compound, which is a drug or is metabolized further to a drug - not just simple cleavage of a group from the prodrug
Protecting Group Analogy for the Concept of Prodrugs Scheme 8.1 analogous to carrier-linked analogous to bioprecursor Keep in mind that a prodrug whose design is based on rat metabolism may not be effective in humans.
Mechanisms of Prodrug ActivationCarrier-Linked Prodrugs Most common activation reaction is hydrolysis.
Ideal Drug Carriers 1. Protect the drug until it reaches the site of action 2. Localize the drug at the site of action 3. Allow for release of drug 4. Minimize host toxicity 5. Are biodegradable, inert, and nonimmunogenic 6. Are easily prepared and inexpensive 7. Are stable in the dosage form
Carrier Linkages for Various Functional GroupsAlcohols, Carboxylic Acids, and Related Groups • Most common prodrug form is an ester • esterases are ubiquitous • can prepare esters with any degree of hydrophilicity or lipophilicity • ester stability can be controlled by appropriate electronic and steric manipulations
Prodrugs for Alcohol-Containing Drugs Table 8.1 Ester analogs as prodrugs can affect lipophilicity or hydrophilicity
To accelerate the hydrolysis rate: • attach an electron-withdrawing group if a base hydrolysis mechanism is important • attach an electron-donating group if an acid hydrolysis mechanism is important • To slow down the hydrolysis rate: • make sterically-hindered esters • make long-chain fatty acid esters
Another Approach to Accelerate HydrolysisIntramolecular hydrolysis of succinate esters Scheme 8.2
Enolic hydroxyl groups can be esterified as well. antirheumatic agent
Maintaining Water Solubility of Carboxylate Prodrugs Can vary pKa by appropriate choice of R and R
Amine Prodrugs Table 8.2 Amides are commonly not used because of stability Activated amides (low basicity amines or amino acids) are effective pKa of amines can be lowered by 3 units by conversion to N-Mannich bases (X = CH2NHCOAr)
N-Mannich base (R = CH2NHCOPh) has a log D7.4 two units greater than the parent compound.
Another approach to lower pKa of amines and make more lipophilic. Imine (Schiff base) prodrug hydrolyze imine and amide to GABA inside brain anticonvulsant
A Reductive Carrier-Linked Prodrug Approach Scheme 8.3
Prodrugs of Sulfonamides A water soluble prodrug of the anti-inflammatory drug valdecoxib (8.9) has been made (8.10).
Prodrug Analogs of Carbonyl Compounds Table 8.3 imines oximes ketals Acetals or ketals can be made for rapid hydrolysis in the acidic medium of the GI tract.
Prodrug Stability Properties Choice of hydrolytic prodrug group: The group must be stable enough in water for a shelf life of > 2 years (13 year half-life), but must be hydrolyzed in vivo with a half-life < 10 minutes. Therefore, in vivo/in vitro lability ratio about 106.
Prodrug for Increased Water Solubility Prodrug forms for aqueous injection or opthalmic use poor water solubility corticosteroid
To avoid formulation of etoposide with detergent, PEG, and EtOH (used to increase water solubility), it has been converted to the phosphate prodrug.
Prodrug for Improved Absorption Through Skin corticosteroids - inflammation, allergic, pruritic skin conditions
Better absorption into cornea for the treatment of glaucoma The cornea has significant esterase activity
Prodrug for Site Specificity Bowel sterilant (administer rectally) Prodrug R = Ac (administered orally) Hydrolyzed in the intestines
Prodrug for Site Specificity The blood-brain barrier prevents hydrophilic molecules from entering the brain, unless actively transported. The anticonvulsant drug vigabatrin (see Chapter 5) crosses poorly. A glyceryl lipid (8.17, R = linolenoyl) containing one GABA ester and one vigabatrin ester was 300 times more potent in vivo than vigabatrin.
Site Specificity Using Enzymes at the Site of Action Phosphatase should release the drug selectively in tumor cells. This approach has not been successful because the prodrugs are too polar, enzyme selectivity is not sufficient, or tumor cell perfusion rate is poor.
Enzyme-Prodrug Therapies For selective activation of prodrugs in tumor cells Two steps: 1. incorporate a prodrug-activating enzyme into a target tumor cell 2. administer a nontoxic prodrug which is a substrate for the exogenous enzyme incorporated
Criteria for Success with Enzyme-Prodrug Therapies The prodrug-activating enzyme is either nonhuman or a human protein expressed poorly 2. The prodrug-activating enzyme must have high catalytic activity 3. The prodrug must be a good substrate for the incorporated enzyme and not for other endogenous enzymes 4. The prodrug must be able to cross tumor cell membranes 5. The prodrug should have low cytotoxicity and the drug high cytotoxicity 6. The activated drug should be highly diffusable to kill neighboring nonexpressing cells (bystander killing effect) 7. The half-life of the active drug is long enough for bystander killing effect but short enough to avoid leaking out of tumor cells
Antibody-Directed Enzyme Prodrug Therapy (ADEPT) An approach for site-specific delivery of cancer drugs. Phase One: An antibody-enzyme conjugate is administered which binds to the surface of the tumor cells. The antibody used has been targeted for the particular tumor cell. The enzyme chosen for the conjugate is one that will be used to cleave the carrier group off of the prodrug administered in the next phase. Phase Two: After the antibody-enzyme has accumulated on the tumor cell and the excess conjugate is cleared from the blood and normal tissues, the prodrug is administered. The enzyme conjugated with the antibody at the tumor cell surface catalyzes the conversion of the prodrug to the drug when it reaches the tumor cell.
ADEPT Advantages: 1. Increased selectivity for targeted cell 2. Each enzyme molecule converts many prodrug molecules 3. The released drug is at the site of action 4. Concentrates the drug at the site of action 5. Demonstrated to be effective at the clinical level Disadvantages: 1. Immunogenicity and rejection of antibody-enzyme conjugate 2. Complexity of the two-phase system and i.v. administration 3. Potential for leakback of the active drug
An example is carboxypeptidase G2 or alkaline phosphatase linked to an antibody to activate a nitrogen mustard prodrug. Scheme 8.4 Humanization of antibodies minimizes immunogenicity. Note the prodrug-activating enzyme is a bacterial enzyme.
Antibody-Directed Abzyme Prodrug Therapy (ADAPT) Instead of using a prodrug-activating enzyme, a humanized prodrug-activating catalytic antibody (abzyme) can be used. Ideally, the abzyme catalyzes a reaction not known to occur in humans, so the only site where the prodrug could be activated is at the tumor cell where the abzyme is bound. Antibody 38C2 catalyzes sequential retro-aldol and retro-Michael reactions not catalyzed by any known human enzyme. Found to be long-lived in vivo, to activate prodrugs selectively, and to kill colon and prostate cancer cells.
Gene-Directed Enzyme Prodrug Therapy (GDEPT)Also known as suicide gene therapy A gene encoding the prodrug-activating enzyme is expressed in target cancer cells under the control of tumor-selective promoters or by viral transfection. These cells activate the prodrug as in ADEPT. Scheme 8.6
Prodrug for Stabilityprotection from first-pass effect Oral administration has lower bioavailability than i.v. injection. antihypertension plasma levels 8 times that with propranolol
Prodrugs for Slow and Prolonged Release 1. To reduce the number and frequency of doses 2. To eliminate night time administration 3. To minimize patient noncompliance 4. To eliminate peaks and valleys of fast release (relieve strain on cells) 5. To reduce toxic levels 6. To reduce GI side effects Long-chain fatty acid esters hydrolyze slowly Intramuscular injection is used also
Sedative/tranquilizer/antipsychotic slow release inject i.m. Antipsychotic activity for about 1 month
Prodrugs to Minimize Toxicity Many of the prodrugs just discussed also have lower toxicity. For example, epinephrine (for glaucoma) has ocular and systemic side effects not found in dipivaloylepinephrine.
Prodrug to Increase Patient Acceptance The antibacterial drug clindamycin (8.28) is bitter and not well tolerated by children. Clindamycin palmitate is not bitter. Either not soluble in saliva or does not bind to the bitter taste receptor or both.
Prodrug to Eliminate Formulation Problems Formaldehyde is a gas with a pungent odor that is used as a disinfectant. Too toxic for direct use. It is a stable solid that decomposes in aqueous acid. The pH of urine in the bladder is about 4.8, so methenamine is used as a urinary tract antiseptic. Has to be enteric coated to prevent hydrolysis in the stomach.
Areas of Improvement for Prodrugs • site specificity • protection of drug from biodegradation • minimization of side effects
Macromolecular Drug Delivery To address these shortcomings, macromolecular drug delivery systems have been developed. A bipartate carrier-linked prodrug in which the drug is attached to a macromolecule, such as a synthetic polymer, protein, lectin, antibody, cell, etc. Absorption/distribution depends on the physicochemical properties of macromolecular carrier, not of the drug. Therefore, attain better targeting. Minimize interactions with other tissues or enzymes. Fewer metabolic problems; increased therapeutic index.
Disadvantages of Macromolecular Delivery Systems • Macromolecules may not be well absorbed • Alternative means of administration may be needed (injection) • Immunogenicity problems
Macromolecular Drug Carriers Synthetic polymers Aspirin linked to poly(vinyl alcohol) has about the same potency as aspirin, but less toxic.
Steric Hindrance by Polymer Carrier poly(methacrylate) to enhance water solubility testosterone No androgenic effect Polymer backbone may be sterically hindering the release of the testosterone.