Dynamic Programming: BLAST
Doug Raiford Lesson 5. Dynamic Programming: BLAST. Left off…. Recursive alignment solution exponential. Number of sub-problems 3 n This is exponential. And so on. Complexity analysis. Fixed: best Linear: next best Polynomial (n 2 ): not bad Exponential (3 n ): very bad Big O notation


Dynamic Programming: BLAST
E N D
Presentation Transcript
Doug Raiford Lesson 5 Dynamic Programming: BLAST
Left off… • Recursive alignment solution exponential Number of sub-problems 3n This is exponential And so on BLAST
Complexity analysis • Fixed: best • Linear: next best • Polynomial (n2): not bad • Exponential (3n): very bad • Big O notation • O(1), O(n), O(n3), O(3n) Big ONotation BLAST
How can improve the complexity of alignment? • If a recursive algorithm solves the same sub-problems over and over… • Just keep track of them, and don’t redo BLAST
Known as dynamic programming • Needleman-Wunsch algorithm • ac__tcg • acagtag • Could maintain a hash of all visited solutions • But there is actually a more elegant solution • Maintain a matrix of scores • Mismatch penalty multiples in first row/column BLAST
Populating the table • Scoring • Match: +1 • Mismatch: 0 • Gap: –1 • Vertical/Horiz. move: Score + (simple) gap penalty • Diagonal move: Score + match/mismatch score • Take the MAX of the three possibilities BLAST
Fill-out rest of table • Fill out row-wise • Scoring • Match: +1 • Mismatch: 0 • Gap: –1 BLAST
Fill-out rest of table • Fill out row-wise • Optimal alignment score in lower right hand corner BLAST
Figure out alignment • Follow the source of “MAX” BLAST
Alignment • = GAP in top sequence • = GAP in left sequence • = ALIGN both positions • One path from the previous table: • Corresponding alignment (start at the end):AC--TCG ACAGTAG BLAST
Practice • Find an optimal alignment for these two sequences: GCGGTT GCGT • Match: +1 • Mismatch: 0 • Gap: –1 BLAST
Practice • Find an optimal alignment for these two sequences: GCGGTT GCGT • Match: +1 • Mismatch: 0 • Gap: –1 GCGGTTGC-GT- Score = +2 BLAST
Summary – pseudo code • F is the scoring matrix, S is a similarity matrix for determining match/mismatch penalties, A and B are the sequences, d is the gap penalty for i=0 to length, (A) F(i,0) ← d*i for j=0 to length(B) F(0,j) ← d*j for i=1 to length(A) for j = 1 to length(B) { Choice1 ← F(i-1,j-1) + S(A(i), B(j)) Choice2 ← F(i-1, j) + d Choice3 ← F(i, j-1) + d F(i,j) ← max(Choice1, Choice2, Choice3) } BLAST
Once the F matrix is computed… AlignmentA ← "" AlignmentB ← "" i ← length(A) j ← length(B) while (i > 0 and j > 0){ Score ← F(i,j) ScoreDiag ← F(i - 1, j - 1) ScoreUp ← F(i, j - 1) ScoreLeft ← F(i - 1, j) if (Score == ScoreDiag + S(A(i-1), B(j-1))){ AlignmentA ← A(i-1) + AlignmentA AlignmentB ← B(j-1) + AlignmentB i ← i - 1 j ← j - 1 } else if(Score == ScoreLeft + d){ AlignmentA ← A(i-1) + AlignmentA AlignmentB ← "-" + AlignmentB i ← i - 1 } otherwise (Score == ScoreUp + d){ AlignmentA ← "-" + AlignmentA AlignmentB ← B(j-1) + AlignmentB j ← j - 1 } } while (i > 0){ AlignmentA ← A(i-1) + AlignmentA AlignmentB ← "-" + AlignmentB i ← i - 1 } while (j > 0){ AlignmentA ← "-" + AlignmentA AlignmentB ← B(j-1) + AlignmentB j ← j - 1 } BLAST
What if one sequence was much longer? • If no penalty for gaps at ends will align optimally with internal sequences atgccgcatactgccgcaggagatcaggactttcatgaatatcatcatgcgtgggagttcag tgccgcaggagatcaggactttca BLAST
Known as semi-global alignment • Global alignment means using the entirety of both sequences • Semi-global alignment allows gaps at the ends for free. • Initialize first row and column to all 0’s • Allow free horizontal/vertical moves in last row and column BLAST
Local alignment • Local alignment – find the best matching subsequence CGATGAAATGGA • This is achieved by allowing a 4th alternative at each position in the table: zero. BLAST
Local alignment • Smith-Waterman algorithm • No negatives • Match: +1 • Mismatch: -1 • Gap: -1 • Find max • Follow path till reach zero BLAST
How complex? • Matrix is MxN • Now polynomial • Have gone from exponential to polynomial • Fixed: best • Linear: next best • Polynomial (n2): not bad • Exponential (3n): very bad O(n2) BLAST
Is this good enough? • If use entire genome sequence as target and perform a local alignment • Genome is approximately 3 billion base-pares • If we have a database of the human genome • Somewhere around 35,000 genes • Would have to do 35,000 global alignments • Must do better! O(n2) not good enough BLAST
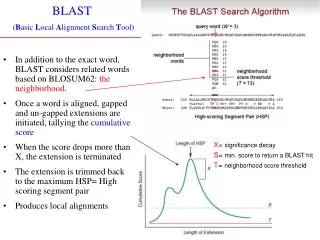
Basic Local Alignment Search Tool • BLAST (Altschulet al. 1990) • Look for areas of interest (linear search) in large string (database) then align just those regions • Can move to near linear time complexity BLAST
Algorithm • Use a sliding window to identify all words (length 3: for proteins or length 11: DNA) in query • Find all locations of these words in the database • Locations where find 2 matches within a certain distance are areas of interest • Align just these areas of interest atgagctatcgctgatgtaccat atgagctatcg tgagctatcgc And so on… gagctatcgct agctatcgctg BLAST
How likely is an 11-mer match? • 411 = 4,194,304 so chance of a random hit: once every 4 million nt’s • Odds of a second hit a short distance away? • Drastically reduced alignment work Now all the way up to linear Fixed: best Linear: next best Polynomial (n2): not bad Exponential (3n): very bad BLAST
What do you give-up? • Way faster (linear) but you miss some possibly important hits • What if there are not two contiguous identical stretches of nucleotides? Speed Sensitivity BLAST
Next lesson… • How improve sensitivity • Also, what if have more than two sequences Sensitivity BLAST
Bit Scores • Scores are affected by sequence lengths • If want scores that can be compared across different query lengths need to normalize • Term “bit” comes from fact that probabilities are stored as log2 values (binary, bit) • Done so can add across length of sequence instead of multiply BLAST