Download
1 / 27
290 likes | 501 Views
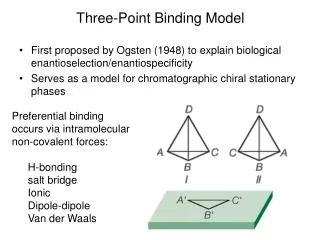
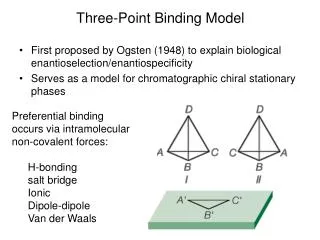
Three-Point Binding Model. First proposed by Ogsten (1948) to explain biological enantioselection/enantiospecificity Serves as a model for chromatographic chiral stationary phases. Preferential binding occurs via intramolecular non-covalent forces: H-bonding salt bridge Ionic Dipole-dipole

E N D
Three-Point Binding Model • First proposed by Ogsten (1948) to explain biological enantioselection/enantiospecificity • Serves as a model for chromatographic chiral stationary phases • Preferential binding occurs via intramolecular non-covalent forces: • H-bonding • salt bridge • Ionic • Dipole-dipole • Van der Waals
Enantioselection by an Enzyme CH2OH moieties are different because of non-equivalent binding sites in the enzyme
Three-Point Binding Model - Enantiospecificity • Only one enantiomer binds to enzyme & is involved in reaction
we get enantiospecificity (substrate & biomolecule are chiral) • To do this efficiently, we need a large biomolecule to align three binding sites to give high specificity • One problem with model: • Model is a static representation → “lock & key”
Binding • The cost of binding: Km (Michaelis constant): small value indicates high affinity for substrate Kbinding ( ~ 1/Km) Strong binding → K > 1 ΔG= -RT ln K ΔG must be –ve
ΔGbinding = ΔHbinding- TΔSbinding For 2 molecules in, 1 out: ΔS is –ve (-TΔS) term is +ve • Entropy disfavors binding of substrate to enzyme • To get good binding, need –ve ΔH (i.e. bond formation) • Each non-covalent interaction is small (H-bond ~ 5 kcal/mol), but still gives a –ve ΔH • Enzymes use many FG’s to sum up many weak non-covalent interactions (i.e. 3 points)
Tyrosyl-tRNA synthase • Use binding to orient CO2- nucleophile adjacent to P specifically as electrophile → specificity • Many non-covalent interactions overcome entropy of binding: H-bonds Can isolate this complex in the absence of tRNA
Bind ATP Binding AAs * 3 point binding enantiospecificity ATP, not dATP Tyr specificity * * *Main chain contacts
Orient PO4 towards CO2- * Increase P+ * * *Main chain contacts
We have examined the crystal structure of tyrosyl-tRNA synthase (Tyr & ATP bound) • Key contacts • 3 point binding model for (S)-tyrosine • We inferred geometry of bound ATP prior to reaction (i.e. ATP is no longer bound to enzyme) Step 1: • CO2- attacks PO42- () giving pentacoordinate P (trigonal bipyramidal) intermediate
Step 2: • Diphosphate must leave • Cannot “see” this step PPi has already left the enzyme site in the crystal structure • However, can use model building to include P & P of ATP: Thr40 & His45 form H-bonds to P **Stronger H-bonds are formed in TS than in trig. Bipyramidal intermediate Lower TS energy accelerate collapse of intermediate Gln195
Tests of Mechanism • Site-directed mutagenesis • Replace Gln195 with Gly (Gln195Gly) • Rate slows by > 1000 fold • ΔΔG ~ 4 kcal/mol • Developing -ve charge (on oxygen) in TS is no longer stabilized • Energy diagram? • Other mutants: • Tyr34Phe • His48Gly • These other mutations showed smaller decreases in ΔG • All contribute in some way to stabilize TS
Do Thr-40 & His-45 really bind / phosphates? Thr 40 Ala ( 7000 fold) His 45 Gly ( 300 fold) Both decelerate the reaction Double mutant 300,000 fold slower!
A Chemical Model for Adenylate Reaction Mimic the proximity effect in an enzyme with small organic molecules: Rate is comparable to tyrosyl-adenylate formation unimolecular reaction Detect by UV
Step 2leads to adenylate; CO2H group is now activated • Once activated, tRNAtyr-OH can bind Step 3: • 3’-OH attacks acyl adenylate • -ve charge increases on O of carbonyl H-bonding stabilizes this charge (more in TS than in SM) • H-bonding (of Gln) is “more important” for TS
X-ray Structure of tRNAGln 3-’OH • Example of tRNA bound to tRNA synthase (stable without Gln) • tRNA (red) binds to enzyme via multiple H-bonds • 3’-OH oriented close to ATP (consistent with proposed mechanism in tyrosyl-tRNA) ATP
Unique Role of Methionine • Recall, Methionine is the 1st amino acid in a peptide/protein (start codon) • As seen previously, Met is also formylated From N-formyltetrahydrofolate protected
Reaction is catalysed by becoming pseud-intramolecular (recall DNA template synthesis): Ribosome holds pieces together Ribosome is cellular “workbench” Protection with formyl group allows condensation one way around only (only one nucleophile) tRNAfMet falls off P site Dipeptide moves over to P site
Control of Sequence • mRNA (messenger RNA) made by copying sequence of DNA in gene • Goes to ribosome, along with rRNA (ribosomal RNA-part of ribosome structure) & tRNA (with AAs attached) • In mRNA, 3 nucleotides of specific sequence encode 1 amino acid (CODON) • R-tRNAR has 3 nucleotides complementary through base pairing to the codon for R • Specific binding at A site • Codons for start & stop control the final protein length
Met Tyr Arg Met Tyr P site CODON A site Rxn & translocation P site Tyr Met A site Arg
Catalysis of Reaction? • Synthesis on ribosome is faster by 107 than rxn without ribosome • Peptide formation is not catalyzed by protein → no protein within 20 Ǻ of “active site” • rRNA (catalytic RNA) has been proposed : Adenosine from rRNA
However, modification of bases has shown little effect on catalytic activity (2-fold decrease) • May be the 2’-OH (of tRNA) at last nucleotide on P site: i.e., the substrate! (see Nature Struct. Mol. Biol. (2004), 11, p 1101 • Modified sugar at 3’OH: • OH → H • OH → F Both substitutions reduce rate by 106!
Why the Reduction in the Rate? P site A site Accounts for most of rate acceleration e.g. of catalytic RNA & substrate catalysis