Lecture 7. Computing Protein Structures
CS882, Fall 2006. Lecture 7. Computing Protein Structures. Current attempts: Threading: RAPTOR Consensus: ACE Fragment assembly Can we compute the protein structures eventually? Your projects. Homologous proteins have similar structure and functions.


Lecture 7. Computing Protein Structures
E N D
Presentation Transcript
CS882, Fall 2006 Lecture 7. Computing Protein Structures • Current attempts: • Threading: RAPTOR • Consensus: ACE • Fragment assembly • Can we compute the protein structures eventually? Your projects.
Homologous proteins have similar structure and functions • Being homologous means that they have evolved from a common ancestral gene. Hence at least in the past they had the same structure and function. • Caution: old genes can be recruited for new functions. Example: a structural protein in eye lens is homologous to an ancient glycolytic enzyme. • Homology search is done by BLAST, or PatternHunter for more sensitivity. BLAST will work with over 30% sequence identity.
Conserving core regions • Homologous proteins usually have conserved core regions. • When we model one protein after a similar protein with known structure, the main problem becomes modeling loop regions. • Modeling loops can also depend on database to some degree. • Side chains: on a few side-chain conformations frequently occur – they are called rotamers, there is a such a database.
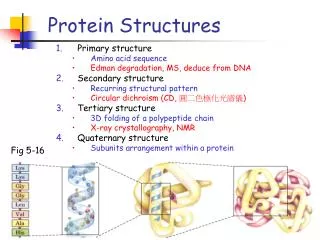
Primary, secondary, and tertiary • There are many secondary structure prediction programs. However, without considering tertiary structure, we will never be correct solely predicting secondary structures. • Most tertiary structure prediction programs today depend on good secondary predictions. This is also not good: you cannot get right tertiary structure with wrong starting information. • They must be done together.
There are not too many candidates! • There are only about 1000 topologically different domain structures. There is no reason whatsoever that we cannot compute their structures accurately. • Ab initio method – we have heard about it. • Another promising method is threading (separate lecture). • After threading, an important step is “refinement”, perhaps by fragment assembly. This will be a separate topic (Xin Gao). • Folding membrane proteins is a quite different topic (Richard Jang). • Now we go to threading.
target sequence • MTYKLILNGKTKGETTTEAVDAATAEKVFQYANDNGVDGEWTYTEtemplate library Protein Threading • Make a structure prediction through finding an optimal placement (threading) of a protein sequence onto each known structure (structural template) • “placement” quality is measured by some statistics-based energy function • best overall “placement” among all templates may give a structure prediction
Introduction to Linear Program • Optimize (Maximize or Minimize) a linear objective function • e.g. 2x+3y+4z • The variables satisfy some linear constraints. e.g. • x+y-z >=1 • 2x+y+3z=3 • integer program (IP) =linear program (LP) + integral variables • LP can be solved within polynomial time --- Interior point method. Simplex method also runs fast. We used IBM package. • Polynomial time for IP not likely, NP-hard • IP can be relaxed to LP, solve the non-integral version • Branch-and-bound or branch-and-cut (may cost exponential time)
Why Integer Programming? • Treat pairwise potentials rigorously • critical for fold-level targets • Existing Exact algorithms for pairwise potentials • High memory requirement, or • Expensive computational time • Exploit correlations between various kinds of item scores in the energy function • 99% real data generate integral solutions directly, no branch-and-bound needed.
Different approaches • Approximation Algorithm • Interaction-Frozen Algorithm (A. Godzik et al.) • Monte Carlo Sampling (T. Madej et al.) • Double dynamic programming (D. Jones et al.) • Recursive dynamic programming (R. Thiele et al.) • Exact Algorithm • Branch-and-bound (R.H. Lathrop et al.) • Exploit the relationship among various scoring parameters, fast self-threading • Divide-and-conquer (Y. Xu et al.) • Exploit the topological structure of template contact graphs
Formulating Protein Threading by LP • Protein Threading Needs: • Construction of Template Library • Design of Energy Function • Sequence-Structure Alignment • Template Selection and Model Construction
Threading Energy Function how preferable to put two particular residues nearby: Ep (Pairwise potential) how well a residue fits a structural environment: Es (Fitness score) sequence similarity between query and template proteins: Em (Mutation score) alignment gap penalty: Eg (gap score) Consistency with the secondary structures: Ess E= Ep + Es + Em + Eg + Ess Minimize E to find a sequence-structure alignment
Contact Graph • Each residue as a vertex • One edge between two residues if their spatial distance is within a given cutoff. • Cores are the most conserved segments in the template: alpha-helix, beta-sheet template
Variables • x(i,l) denotes core i is aligned to sequence position l • y(i,l,j,k) denotes that core i is aligned to position l and core j is aligned to position k at the same time.
Formulation 1 Eg , Ep Es , Ess , Em Encodes scoring system Encodes interaction structures: the first makes sure no crosses; the second is quadratic, but can be converted to linear: a=bc is eqivalent to: a≤b, a≤c, a≥b+c-1
Formulation used in RAPTOR Eg, Ep Es, Ess, En Encodes scoring system Encodes interaction structures
Solving the Problem Practically • More than 99% threading instances can be solved directly by linear programming, the rest can be solved by branch-and-bound with only several branch nodes • Less memory consumption • Less computational time • Easy to extend to incorporate other constraints
Fold Recognition • Support Vector Machines (SVM) Approach • Features are extracted from the alignments • A threading pair is treated as a positive pattern only if they are in at least fold-level similarity • 60,000 threading pairs are employed to train SVM model. • 5% more targets are recognized by SVM approach than the traditional z-Score
Target Category Hard Easy Prediction Difficulty CM: Comparative Modelling, HM: Homology Modelling FR: Fold Recogniton, NF: New Fold
Lindahl Benchmark Test 976*975 threading pairs are tested, the results of other servers are taken from Shi et al.’s paper.
LiveBench Test LiveBench 6 LiveBench 7 (http://bioinfo.pl/LiveBench)
CASP5/CAFASP3 • 62 targets • Time allowed for each target: • Individual Servers: 48 hours • Meta Servers: 48 hours • Predictors: computer program, no manual intervention (CAFASP3) • Evaluated by computer program • RAPTOR was voted by CASP5 attendees as the most novel approach, at http://forcasp.org CAFASP3: The Third Critical Assessment of Fully Automated Structure Prediction
CAFASP3 Evaluation Criteria • Model • Only the first submission considered for each target, • each server can submit 10 models for each target, • MaxSub (evaluation program) • Superimpose the predicted structure with the experimental structure • Calculate the length of maximum superimposable subsegment within 5Å RMSD • one prediction is regarded as correct only if the length is above a given value.
CAFASP3 Evaluation Criteria • Sensitivity (N-1 Rule) • One miss allowed for each server, i.e., the first models of N-1 out of N targets ranked • Specificity • Rank the first models of all targets according to their zScores • S(M): # Correct before the first M false positives • Average of S(1),S(2),…,S(5)
Specificity Example S(1)=2 S(2)=3 First false positive Second false positive
Sensitivity on FR targets (1) 30 FR targets 54 servers (http://ww.cs.bgu.ac.il/~dfischer/CAFASP3, released on Dec., 2002.)
Sensitivity on FR targets (2) • RAPTOR is weak at recognizing FR(A) targets (need improvement ) • RAPTOR cannot deal with NF targets at all (normal)
Specificity of Servers Out of 33 Targets
CAFASP3 Example • Target ID: T0136_1 • Target Size:144 • Superimposable size within 5Å: 118 • RMSD:1.9Å Red: Experimental Structure Blue/green: RAPTOR model
CASP6, T0199-2, ACE buffalo rank: 9thFrom RAPTOR rank 1 model. TM=0.4183 MaxSub=0.2857. Good parts: 116-134, 286-332 Left: predicted structure. Right: experimental structure
CASP6, T0203 ACE buffalo rank: 1stFrom RAPTOR 2nd model. TM=0.6041, MaxSub=0.3485. Good parts: 19-57, 89-94, 139-178, 224-239, 312-372 RAPTOR first Model ranks 5th Predicted Experimental
CASP6, T0262-2, ACE buffalo rank: 4thFrom Fugue3 6th model. TM=0.4306, MaxSub=0.3459. Good parts: 162-203 Fugue’s top model ranks low Predicted Experimental
CASP6, T0242, NF, ACE buffalo rank: 1From RAPTOR rank 5 model.TM score=0.2784, MaxSub score=0.1645 However, RAPTOR top model ranks 44th ! Trivial error? Predicted Experimental
CASP6, T0238, NF ACE buffalo rank 1stFrom RAPTOR 8th model TM=0.2748, MaxSub=0.1633Good part: 188-237. High TM score, low MaxSub Raptor top model ranks 4th Predicted Experimental
About RAPTOR • Jinbo Xu’s Ph.D. thesis work. • The RAPTOR system has benefited significantly from PROSPECT (Ying Xu, Dong Xu, et al). • Currently distributed by BSI. • References:J. Xu, M. Li, D. Kim, Y. Xu, Journal of Bioinformatics and Computational Biology, 1:1(2003), 95-118. J. Xu, M. Li, PROTEINS: Structure, Function, and Genetics, CASP5 special issue.