Download

1 / 23
230 likes | 370 Views
This research presents a novel method for reverse engineering gene networks at a genome-wide scale, utilizing singular value decomposition (SVD) and robust regression. The approach can effectively work with small datasets without prior knowledge of gene interactions. By applying external stimuli and measuring mRNA concentrations, the algorithm aims to deduce gene connectivity matrices with minimal experimental data (M ≈ O(log(N))). The advantages include reduced data requirements and ease of parallelization, though it may be less efficient for smaller networks. The method is also applicable to protein networks, expanding its potential usage in systems biology.

E N D
Reverse engineering gene networks using singular value decomposition and robust regression M.K.Stephen Yeung Jesper Tegner James J. Collins
General idea Reverse-engineer: • Genome-wide scale • Small amount of data • No prior knowledge • Using SVD for a family of possible solutions • Using robust regression to choose from them
If the system is near a steady state, dynamics can be approximated by linear system of N ODEs: xi = concentration of mRNA (reflects expression level of genes) λi = self-degradation rates bi = external stimuli ξi = noise Wij = type and strength of effect of jth gene on ith gene
Suppositions made: • No time-dependency in connections (so W is not time-dependent), and they are not changed by the tests • System near steady state • Noise will be discarded, so exact measurements are assumed • can be calculated exactly enough
In M experiments with N genes, • each time apply stimuli (b1,…,bN) to the genes • measure concentrations of N mRNAs (x1,…,xN) using a microarray You get: subscript i = mRNA number superscript j = experiment number
Goal is to use as few measurements as possible. By this method (with exact measurements): M = O(log(N)) e.g. in 1st test, the results will be:
System becomes: With A = W + diag(-λi) Compute by using several measurements of the data for X. (e.g. using interpolation) Goal = deduce W (or A) from the rest If M=N, compute (XT)-1, but mostly M << N (this is our goal: M = log(N))
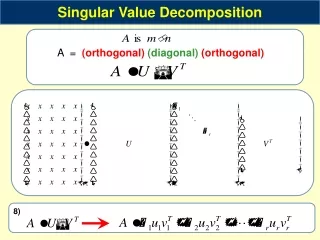
Therefore, use SVD (to find least squares sol.): Here, U and V are orthogonal (UT = U-1) and W is diag(w1,…,wN) with wi the singular values of X Suppose all wi= 0 are in the beginning, so wi= 0 for i = 1…L and wi≠ 0 (i=L+1...L+N)
Then the least squares (L2) solution to the problem is: With 1/wj replaced by 0 if wj = 0 So this formula tries to match every datapoint as closely as possible to the solution.
But all possible solutions are: with C = (cij)NxNwhere cij = 0 if j > L and otherwise just a scalar coefficient How to choose from the family of solutions ? The least squares method tries to match every datapoint as closely as possible → a not-so-sparse matrix with a lot of small entries.
Basing on prior biological knowledge,impose this on the solutions.e.g.: when we know 2 genes are related,the solution must reflect this in the matrix • Work from the assumption that normal gene networks are sparse, and look for the matrix that is most sparsethus: search cij to maximize the number of zero-entries in A
So: • get as much zero-entries as you can • therefore get a sparse matrix • the non-zero entries form the connections • fit as much measurements as you can, exactly: “robust regression” (So you suppose exact measurements)
Do this using L1 regression. Thus, when considering we want to “minimize” A. The L1 regression idea is then to look for the solution C where is minimal. This causes as many zeros as possible. Implementation was done using the simplex method (linear adjustment method)
Thus, to reverse-engineer a network of N genes, we “only” need Mc = O(logN) experiments. Then Mc << N, and the computational cost will be O(N4) (Brute-force methods would have a cost of O(N!/(k!(N-k)!)) with k non-zero entries)
Test 1 • Create random connectivity matrix:for each row, select k entries to be non-zero - k < kmax << N (to impose sparseness) - non-zero entry random from uniform distrib. • Do random perturbations • Do measurements while system relaxes back to its previous steady state → X • Compute by interpolation • Do this M times
Test 1 • Then apply algorithm to become approximation of A • Computed error (with the computed A):
Results: Mc = O(log(N)) • Better than only SVD, without regression:
Test 2 • One-dimensional cascade of genes • Result for N = 400: Mc = 70
Test 3 • Large sparse gene network, with ran- dom connections, external stimuli,… • Results the same as in previous tests
Discussion Advantages: • Very few data needed, in comparison with neural networks, Bayesian models • No prior knowledge needed • Easy to parallelize, as it recovers the connectivity matrix row by row (gene by gene) • Also applicable to protein networks
Discussion Disadvantages: • Less efficient for small networks (M≈N) • No quantification yet of the necessary “sparseness”, though avg. 10 connections is good for a network containing > 200 genes • Uncertain • Especially useful with exact data, which we don’t have
Improvements • Other algorithms to impose sparseness: alternatives are possible both for L1 (basic criterion) as for simplex (implementation) • By using a deterministic linear system of ODEs, a lot has been neglected (noise, time delays, nonlinearities) • Connections could change by experiments; then the use of time-dependent W is necessary