Download

1 / 23
230 likes | 373 Views
A New Set of Multilevel Methods for Obtaining Accurate Energies on the Potential Energy Surface. Wei-Ping Hu ( 胡維平 ) Department of Chemistry and Biochemistry National Chung Cheng University. I. Introduction. H full Y God = E exact Y God Y (FCI/CBS) = Y (FCC/CBS) = Y God

E N D
A New Set of Multilevel Methods for Obtaining Accurate Energies on the Potential Energy Surface Wei-Ping Hu (胡維平) Department of Chemistry and Biochemistry National Chung Cheng University
I. Introduction HfullYGod = EexactYGod Y(FCI/CBS) = Y(FCC/CBS) = YGod E (FCI/CBS) = E (FCC/CBS) = Eexact QCISD(T), CCSD(T) Scale as N 7 Multilevel Methods:EexactE(LL/IBS) + DEHLC + DECB
Currently Used Multilevel Methods: G2, G3, CBS, SAC, G3S, MCG3 HF, MP2, MP4, QCISD(T), empirical HLC 6-31G(d), 6-311G(d,p), 6-311+G(d,p) 6-311+G(2df,p), 6-311+G(3df,2p), G3Large Target Accuracy: QCISD(T)/6-311+G(3df,2p), QCISD(T)/G3Large New Method: (no empirical parameters, Dynamics Calc.) Target Accuracy:QCISD(T)/aug-cc-pVT(Q)Z Dunning’s Corrlation Consistent Basis Sets Basis set extension: MP2, extrapolation
簡稱 • QCISD(T)/cc-pVDZ方法 : QT2 • QCISD(T)/cc-pVTZ方法: QT3 • 估計 QCISD(T)/cc-pVTZ能量之方法 :EQT3 • 估計 QCISD(T)/cc-pVQZ能量之方法 : EQT4 • 估計 QCISD(T)/cc-pVnZ能量之方法 : EQTn • 估計 QCISD(T)/aug-cc-pVTZ能量之方法 : E(A)QT3+ • 估計 QCISD(T)/aug-cc-pVQZ能量之方法 : E(A)QT4+
II. Methods EQT3+ = EQT3 +DE+ EQT3 = QT2 +DETZ DETZ = MP2/cc-pVTZ - MP2/cc-pVDZ DEQZ = MP2/cc-pVQZ - MP2/cc-pVTZ DE+ = MP2/aug-cc-pVTZ - MP2/cc-pVTZ EQT4+ = EQT4 +DE+ EQT4 = EQT3 +DEQZ AQT3+ = QT3 +DE+ AQT4+ = AQT3+ +DEQZ
E(A)QTn 方法 EQTn = -76.376 (H2O) f (n) = ae-bn + c 以外插的方法估計到無窮大basis set時的能量。以QT2(n = 2), EQT3(n = 3), EQT4(n = 4)來估計EQTn的能量[ EQTn = c , as n = ∞ ]。
III. Results and Discussion • EQT4 及EQTn 所需的計算時間相同,但 EQTn因利用外插的方法使基底函數趨近於無窮大,而使其RMSE 較 EQT4 下降了0.6kcal/mol 左右。 • aJ. Phys. Chem.1999, 103, 3139 • 在本研究中EQTn 方法準確度與CCSD(T)-SAC/pTZ 相當,而其所需計算時間較 CCSD(T)-SAC/pTZ方法少。 Table 1 : 原子化能量之誤差(kcal/mol)
在原子化能量計算中我們可得知,single-level中以MP2/cc-pVTZ方法所得的MUE及RMSE都比以MP2/cc-pVDZ方法所得的約小14.0kcal/mol左右,而QT2方法並沒有比MP2方法好。在原子化能量計算中我們可得知,single-level中以MP2/cc-pVTZ方法所得的MUE及RMSE都比以MP2/cc-pVDZ方法所得的約小14.0kcal/mol左右,而QT2方法並沒有比MP2方法好。 • 在multi-level中基底函數為triple-zeta時,擴散函數的貢獻相當重要,而基底函數為quadruple-zeta時,擴散函數的貢獻較少。
Table 2: 文獻上記載之反應能障最佳估計值(kcal/mol) J. Phys. Chem. A. 2001, 105, 2936
以EQT3+,EQT4, EQT4+ 及EQTn 等四種方法所估計出來的DV≠值與文獻上認為最準確的值比較其 MUE 都在1.0 kcal/mol以下,而RMSE 也都約1.0 kcal/mol左右,table 2 其誤差範圍在1.0 kcal/mol 左右,所以本研究中的multilevel 方法以及文獻上的G3S 和MMCG3方法預測反應能障的準確度都相當高。 Table 3 : 能量障礙之誤差(kcal/mol) • G3S 是 Donald G. Truhlar 等人發明的方法,其將 G3 方法中的 HLC 修正省略,而加入半經驗參數來加以校正,而 Larry A. Curtiss 等人發明的 MMCG3 方法與 G3S 類似。
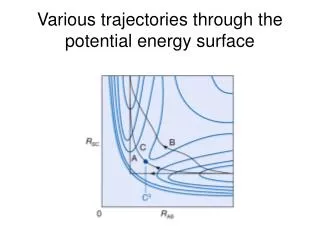
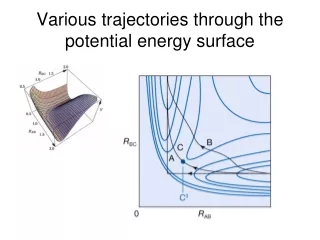
使用multi-level進行位能曲線的計算 我們使用EQT3+、EQT4+ 、 EQTn 、 AQT4+ 、AQTn、PDG2、G3S與MMCG3等multi-level方法計算H2+H、OH+H2、CH4+OH、NH3+OH、以及H2S+H之位能曲線,比較各種方法所得之能量障礙與能障半高寬(S1/2) ,並與文獻值比較。以上五種反應路徑之幾何結構計算是使用: H2+H OH+H2 CH4+OH NH3+OH MP2/6-311+G** H2S+H QCISD(T)/6-311+G**
我們使用的multi-level方法所算出來的ΔV‡與文獻值的誤 差約在0.1~1.9kcal/mol左右,幾乎都比G2與G3系列的方 法良好。 • 實際使用single-level的結果與我們所使用之方法的結果很 接近,相差大約0.2~1.5kcal/mol左右。 • ΔV‡會隨著計算level提升、加大basis set,以及加入diffuse function而降低﹔加入diffuse function的下降量則特 別顯著。 • 在S1/2方面,加大basis set的改變與實際做single-level的趨 勢相同,但是加入diffuse function之後卻呈現不規則變化。 造成這種情況的原因是由於MP2與QCISD(T)兩種計算level在計算位能曲線時,有不同的趨勢所導致。
感 謝 國科會對本研究計畫的經費補助 李宗憓、牟君浩 中國化學會 國立中正大學化學暨生物化學系