Geochemical Kinetics
Geochemical Kinetics. Look at 3 levels of chemical change: Phenomenological or observational Measurement of reaction rates and interpretation of data in terms of rate laws based on mass action Mechanistic


Geochemical Kinetics
E N D
Presentation Transcript
Geochemical Kinetics • Look at 3 levels of chemical change: • Phenomenological or observational • Measurement of reaction rates and interpretation of data in terms of rate laws based on mass action • Mechanistic • Elucidation of reaction mechanisms = the ‘elementary’ steps describing parts of a reaction sequence (or pathway) • Statistical Mechanical • Concerned with the details of mechanisms energetics of molecular approach, transition states, and bond breaking/formation
Reactions and Kinetics • Elementary reactions are those that represent the EXACT reaction, there are NO steps between product and reactant in between what is represented • Overall Reactions represent the beginning and final product, but do NOT include one or more steps in between. FeS2 + 7/2 O2 + H2O Fe2+ + 2 SO42- + 2 H+ 2 NaAlSi3O8 + 9 H2O + 2 H+ Al2Si2O5(OH)4 + 2 Na+ + 4 H4SiO4
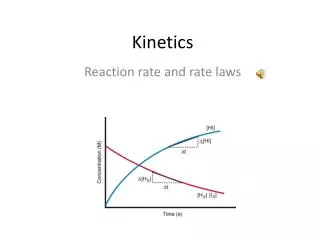
Extent of Reaction • In it’s most general representation, we can discuss a reaction rate as a function of the extent of reaction: Rate = dξ/Vdt where ξ (small ‘chi’) is the extent of rxn, V is the volume of the system and t is time Normalized to concentration and stoichiometry: rate = dni/viVdt = d[Ci]/vidt where n is # moles, v is stoichiometric coefficient, and C is molar concentration of species i
Rate Law • For any reaction: X Y + Z • We can write the general rate law: Rate = change in concentration of X with time, t Order of reaction Rate Constant Concentration of X
Reaction Order • ONLY for elementary reactions is reaction order tied to the reaction • The molecularity of an elementary reaction is determined by the number of reacting species: mostly uni- or bi-molecular rxns • Overall reactions need not have integral reaction orders – fractional components are common, even zero is possible
First step in evaluating rate data is to graphically interpret the order of rxn • Zeroth order: rate does not change with lower concentration • First, second orders: Rate changes as a function of concentration
Zero Order • Rate independent of the reactant or product concentrations • Dissolution of quartz is an example: SiO2(qtz) + 2 H2O H4SiO4(aq) log k- (s-1) = 0.707 – 2598/T
First Order • Rate is dependent on concentration of a reactant or product • Pyrite oxidation, sulfate reduction are examples
First Order Find order from log[A]t vs t plot Slope=-0.434k k = -(1/0.434)(slope) = -2.3(slope) k is in units of: time-1
1st-order Half-life • Time required for one-half of the initial reactant to react
Second Order • Rate is dependent on two reactants or products (bimolecular for elementary rxn): • Fe2+ oxidation is an example: Fe2+ + ¼ O2 + H+ Fe3+ + ½ H2O
2nd Order • For a bimolecular reaction: A+B products [A]0 and [B]0 are constant, so a plot of log [A]/[B] vs t yields a straight line where slope = k2 (when A=B) or = k2([A]0-[B]0)/2.3 (when A≠B)
Pseudo- 1nd Order • For a bimolecular reaction: A+B products If [A]0 or [B]0 are held constant, the equation above reduces to: SO – as A changes B does not, reducing to a constant in the reaction: plots as a first-order reaction
2nd order Half-life • Half-lives tougher to quantify if A≠B for 2nd order reaction kinetics – but if A=B: If one reactant (B) is kept constant (pseudo-1st order rxns):
3rd order Kinetics • Ternary molecular reactions are more rare, but catalytic reactions do need a 3rd component…
Zero order reaction • NOT possible for elementary reactions • Common for overall processes – independent of any quantity measured [A]0-[A]=kt
Pathways • For an overall reaction, one or a few (for more complex overall reactions) elementary reactions can be rate limiting Reaction of A to P rate determined by slowest reaction in between If more than 1 reaction possible at any intermediate point, the faster of those 2 determines the pathway
Initial Rate, first order rxn example • For the example below, let’s determine the order of reaction A + B C • Next, let’s solve the appropriate rate law for k
Rate Limiting Reactions • For an overall reaction, one or a few (for more complex overall reactions) elementary reactions will be rate limiting Reaction of A to P rate determined by slowest reaction in between If more than 1 reaction possible at any intermediate point, the faster of those 2 determines the pathway
Activation Energy, EA • Energy required for two atoms or molecules to react
Transition State Theory • The activation energy corresponds to the energy of a complex intermediate between product and reactant, an activated complex A + B ↔ C± ↔ AB It can be derived that EA = RT + DHC±
Collision Theory • collision theory is based on kinetic theory and supposes that particles must collide with both the correct orientation and with sufficient kinetic energy if the reactants are to be converted into products. • The minimum kinetic energy required in a collision by reactant molecules to form product is called the activation energy, Ea. • The proportion of reactant molecules that collide with a kinetic energy that is at least equal to the activation energy increases rapidly as the temperature increases.
T dependence on k • Svante Arrhenius, in 1889, defined the relationship between the rate constant, k, the activation energy, EA, and temperature in Kelvins: or: Where A is a constant called the frequency factor, and e–EA/RT is the Boltzmann factor, fraction of atoms that aquire the energy to clear the activation energy
Arrhenius Equation y = mx + b Plot values of k at different temperatures: log k vs 1/T slope is EA/2.303R to get activation energy, EA
Activation Energy • EA can be used as a general indicator of a reaction mechanism or process (rate-limiting)