Download

1 / 55
570 likes | 1.23k Views
Neuromuscular Emergencies. July 28, 2010 Sandra Derghazarian. Outline. Approach to rapidly progressing LMN weakness Myasthenic crisis GBS. Intro. Three major tasks: Assess stability, signs of imminent resp failure Generate differential diagnosis Knowledge of localisation

E N D
Neuromuscular Emergencies July 28, 2010 Sandra Derghazarian
Outline • Approach to rapidly progressing LMN weakness • Myasthenic crisis • GBS
Intro • Three major tasks: • Assess stability, signs of imminent resp failure • Generate differential diagnosis • Knowledge of localisation • Knowledge of major disorders • Determine management
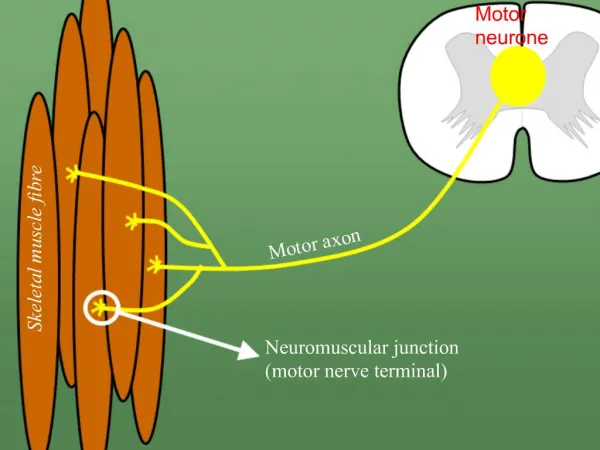
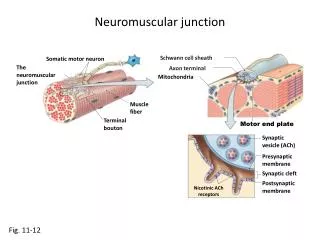
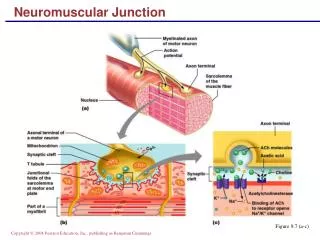
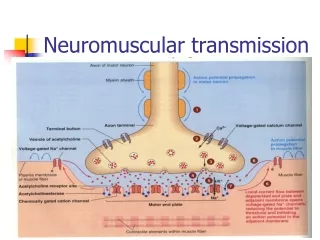
Brief Review of Anatomy • Motor unit anatomy • Anterior horn cell • Its motor axon • The synaptic cleft • The muscle fibers it innervates • Remember sensory and autonomic systems • Many disorder with rapidly progressing peripheral neuropathic weakness will have sensory loss and dysautonomia
Classification • Motor neuron • Loss of anterior horn cells • Motor axon • Disruption of myelin sheath • Axonal degeneration • Neuromuscular junction • Pre-synaptic (e.g. Release of Ach) • Post-synaptic (e.g. Abnormalities of Ach receptor) • Muscle • Membrane, contractile elements • Genetic or acquired secondary (infex, inflamm, vasc.)
Approach to Determining Cause of Weakness • Knowledge of some of the possible disorders • Focused history • Physical exam • Lab studies
History – Key Elements • Pre-existing NM disorder? • MG – exacerbation 2ary systemic illness, medication • ALS – accelerated phase, decompensation 2ary pneumonia • Pre-existing systemic disorder? • Malignancy, CTD, sepsis • What drugs is pt taking? • Any recent illness? • Diet in last 48 hours? • Shellfish (saxitoxin, brevetoxin) • Home-canned goods (botulinum toxin) • Any possible exposure to tick bite, snake bite? • Any sensory or autonomic symptoms?
Drugs • Diuretics • Hypokalemia • Corticosteroids, statins, colchicine, cyclosporine, cocaine, chloroquine, L-tryptophan, penicillamine, zidovudine • Myotoxic effect • Amiodarone, cytarabine, streptokinase • Demyelinating neuropathy • Magnesium-containing antacids with pre-existing renal insufficiency • Hypermagnesemia
Physical Exam • Vital signs • Unstable? • Dysautonomia? • Respiratory status
Signs of impending NM resp failure • Tachypnea, sinus tachycardia • Staccato speech • Inability to count to 20 • Profound weakness of neck flexion • Use of accessory muscles (visible, palpable) • Orthopnea • Paradoxical breathing pattern • Signs of bulbar dysfunction (nasal voice, accumulation of saliva, weak cough) • think about aspiration!
Physical Exam • Vital signs • Note any dysautonomia • Presence and pattern muscle weakness • Proximal (myopathy), distal (peripheral neuropathy) • Symmetric, asymmetric • Involvement of cranial muscles • Reflexes • Sensory changes • Dysautonomia
Localisation of the Disorders • Clinical picture varies depending on which part of motor unit is involved
Regroup • Is respiratory failure imminent? • Should ICU be involved? • Where can I localise motor findings? • Does it fit with sensory findings? • Does it fit with autonomic findings? • Does it fit with the history? • Can the history help me narrow things down?
Laboratory Studies • CBC • Anemia or leukocytosis - systemic disease • Eosinophilia - possibly elevated in vasculitic neuropathy, porphyria • Lytes, Cr, BUN, Ca, Mg, PO4, • Liver enzymes (consider EtOHmyotoxicity) • CK (myopathy if very elevated) • ESR (infectious or inflammatory disorders) • CXR (pneumonia, atelectasis, elevated hemidiaphragm) • EKG • Changes associated with electrolyte imbalances • Arrythmias 2ary to dysautonomia in GBS • Axis deviation – may be suggestive of cardiomyopathy
Assess Respiratory Status • Tests in ER • MIP • MEP • FVC • ABG • 20/30/40 rule • VC: 20 ml/kg • MIP: -30cmH20 • MEP: +40cm H20
Myasthenia Gravis • Disorder of transmission across NM junction • Auto-immune and congenital form • Epidemiology (auto-immune form): • 200-400 cases per million population • Women > men (3:2) • Bimodal incidence • F: 20s, 30s; M: 50s, 60s • 5-10% co-association with other auto-immune disorders
Classification • Auto-immune – two forms • Acquired anti-AChR Abs (85%) • Acquired anti-MuSK Abs, a muscle-specific TK • 40-50% of anti-AChR seronegative pts • Congenital • Heterogeneous group (pre or post-synaptic) • Of note: do not affect respiratory muscles therefore do not present with myasthenic crisis
Clinical Features • Painless, fatigable weakness of voluntary muscles • Repeated activity progressive paresis • Rest restoration of strength (at least partial) • Usually insidious onset • May occur more rapidly after precipitant (stress, infection) • Association with thymic abnormalities • 10-15% thymoma • 50-70% thymic lymphoid hyperplasia
Clinical Features • Presenting symptoms: • MuSK-MG • Younger women • Predominantly facial, bulbar and respiratory weakness • Relatively mild limb weakness
Myasthenic Crisis • Myasthenic weakness leading to respiratory failure and need for ventilatory assistance • Severe weakness of respiratory muscles and/or • Severe weakness of upper airway muscles (bulbar myasthenia)
Prevalence and Characteristics • Life-time prevalence: 20-30% • Early onset younger pt, median onset w/in 8 mos, fast recovery • Late onset older pts, later in dz course, slower recovery • White pts respond more poorly than black pts • Pregnancy aggravates MG in 30% of women • High potential mortality of crisis
Precipitants • Elements to look for in history/chart: • Poor control of generalised disease • Medical treatment for bulbar myasthenia • Steroids and anti-cholinesterases • Use of certain drugs (next slide) • Systemic infection, esp. of respiratory tract • Aspiration • Surgery • Others (in refractory myasthenia): • Emotional stress • Hot environment • Hyperthyroidism
Drugs • Anticholinesterases can also lead to myasthenic crisis • Signs of excessive cholinergic activity • Miosis, diarrhea, salivation, abdominal cramps, sweating, weakness
Investigations • CBC, extended lytes, BUN, Cr, liver enzymes • CXR, U/A +/- blood cultures • Obtain VC, MIP, MEP – 20/30/40 rule
Investigations • Repetitive motor nerve stimulation • Stimulate motor nerve at 2-3 Hz and measure CMAP of stimulated muscle • Positive if >er 10% decrement in amplitude of CMAP from the 1st to the 5th potential • Positive in about 75% of patients withgeneralized MG, if • Proximal & clinicallyinvolved muscles are tested • Muscle is warm • More than one muscle istested • Single fibre EMG • Tensilon test not recommended in pt suspected of being in crisis • False postive, false negative • Risk of worsening muscle weakness in pts with anticholinesterase overdose • Worsening of bulbar and respiratory symptoms in MuSK-MG
Management • Monitoring of respiratory status • Recognition of impending resp failure • Tachypnea, inability to count to 20, saliva pooling, nasal voice, NF weakness, paradoxical breathing • Deciding when to intubate • (Code status) • 20/30/40 rule • If in doubt, intubate • ?BiPAP • Limited experience. May reduce prolonged intubatn and trach
Management • General • Stop any meds that may be contributing • Treat any infection • Specific • PLEX and IVIG comparable efficacy • Based on clinical evidence, few RTCs • Earlier response seen with PLEX • More likely to extubate at 14 days, better 1-month functional outcome (Qureshi, et al. Neurology, 1999).
Management • PLEX • Removal of anti AChR and antiMuSK Abs • 1 session/day x 5 • No superiority of PLEX qd x 5 vs qod x 5 • Rapid onset of action (3-10 days) • Need central line with associated complications • PTX, hemorrhage, line sepsis • Caution in pts with sepsis, hypotension; may lead to increased bleeding and cardiac arrhythmias
Management • IVIG • 0.4gm/kg/day x 5 days • Easilyadministered and widelyavailable • Long duration of action • May last as long as 30 days • Side effects • Anaphylaxis in IgA deficiency • Renal failure, pulmonary edema • Aseptic meningitis • Thrombotic complications and stroke
MG – Overall Treatment Summary • 1. Mildweakness: cholinesteraseinhibitors • 2. Moderate-markedlocalized or generalizedweakness • Cholinesteraseinhibitors, and • Thymectomy for patients underage 50-60 yrs • 3. Symptomsuncontrolled on cholinesteraseinhibitors • Prednisone if severe or urgent • Azathioprine • Prednisonefailure • Excessive prednisoneside-effects • 4. Plasma exchange or IV Ig • Impendingcrisis; crisis • Pre-operativeboost • Chronicdiseaserefractory to drugtherapy • 5. If abovefails • Search for residual thymus tissue • Cyclosporine or mycophenylatemofetil (Sem. Neurol., 2001;21:425-440)
GBS • Most common cause of acute and subacute generalised paralysis • Incidence of 0.4 to 1.7/100 000 per yr • Worldwide, all ages, both sexes • Preceding mild resp or GI infection in 60% (1-3 wks) • Campylobacter jejuni (26%), • CMV, EBV, VZV • Influenza, cocksackie, hepatitis A and B, HIV • May also be preceded by: • Surgery • Immunisations
Typical Symptoms & Signs • Sensory • Paresthesias and slight numbness distally earliest Sx • Reduced proprioception and vibration sense (1 wk) • Motor • Weakness • Evolves symmetrically over days to 1-2 weeks • Usually LE before UE, proximal + distal • +/- trunk, intercostal, neck, cranial muscles • Progresses to total motor paralysis and respiratory failure in 5% of cases
Typical Symptoms & Signs • Reflexes • Reduced and then absent • Autonomic dysregulation • Sinus tachycardia/bradycardia, facial flushing, labile BP, excess or loss of sweating, urinary retention • Usually do not persist for >er 1 wk • Other • Myalgias (50%) in hips, thighs, back
Variants • Fisher syndrome • Ophthalmoplegia, ataxia, areflexia • +/- bilateral facial nerve paresis • Associated with anti-GQ1b Ab • Acute motor sensory axonal neuropathy (5% of GBS cases) • Severe and diffuse axonal damage • Abrupt and explosive onset • Severe paralysis, minor sensory features • Slow and poor recovery • Pandysautonomia • Severe orthostatic hypotension, anhidrosis, dry eyes and mouth, fixed pupils, arrhythmia, bowel/bladder dysfunction • Areflexia without somatic motor/sensory involvement • Other variants: • Initial cervico-brachial-pharyngeal muscle involvement • Generalised ataxia without dysarthria or nystagmus • Facial and abducens weakness, distal paresthesias, proximal leg weakness
Laboratory Findings • Most important: CSF, EMG • CSF • Normal pressure • Protein • Early (1st 2 days): Usually normal (>85%) • Later: High (66% in 1st week, 82% in 2nd week) • Amount not correlated with clinical course or prognosis • Acellular or few lymphocytes • 10% : 10-50 lymphocytes, decreases over 2-3 days; if not: other Dx • Oligoclonal bands (10-30%)
Laboratory Findings • EMG • Abnormalities seen within first week of sx • Reduction in motor amplitude • Slowed conduction velocities • Conduction block in motor nerves • Prolonged distal latencies (distal conduction block) • Prolonged/absent F-responses (involvement of proximal parts of nerves and roots)
Laboratory Findings • Hematology • Abnormal only with infection or other disorder • Biochemistry • Mild-severe SIADH in 7-26% • Liver enzymes • Elevated <10% reflecting CMV or EBV infection • ESR: Normal unless co-existing process
Diagnostic Criteria • National Institute of Neurological Disorders and Stroke (NINDS) criteria are based on expert consensus. • Required features include: • Progressive weakness of more than one limb, ranging from minimal weakness of the legs to total paralysis of all four limbs, the trunk, bulbar and facial muscles, and external ophthalmoplegia • Areflexia. While universal areflexia is typical, distal areflexia with hyporeflexia at the knees and biceps will suffice if other features are consistent. • Supportive features include: • Progression of symptoms over days to four weeks • Relative symmetry • Mild sensory symptoms or signs • Cranial nerve involvement, especially bilateral facial nerve weakness • Recovery starting two to four weeks after progression halts • Autonomic dysfunction • No fever at the onset • Elevated protein in CSF with a cell count <10 mm3 • Electrodiagnostic abnormalities consistent with GBS
Differential Diagnosis • Features suggesting another diagnosis: • Sensory level, severe bladder or bowel dysfunction Spinal cord syndrome • Marked asymmetry Mononeuritis multiplex/vasculitis • CSF pleocytosis Infectious disorders: viral, HIV, lyme, poliomyelitis • Very slow nerve conduction velocities, multiple relapses or chronic course -> CIDP • Persistent abdominal pain and psychiatric signs Acute intermittent porphyria
Management • General: • Recommend admission for observation • Can deteriorate rapidly in first days of presentation • M&M: Respiratory failure, dysautonomia • 25% will require mechanical ventilation • Respiratory • Measure MIP/MEP/FVC • Decision to intubate should be based on downward trend • Other measures of respiratory status same • Counting to 20, strength of NF
Management • Dysautonomia • 10% develop hypotension • Volume, +/- pressors • Hypertension • IV labetolol • Other complications • Adynamicileus • PE • Aspiration
Management • PLEX and IVIG • No difference in efficacy between the two • Indications for prompt initiation • Respiratory failure • Bulbar involvement • Inability to walk without assistance • Usually see these signs day 5-10 • May occur anywhere from day 1 – week 3 • Steroids • No proven benefit