Download

1 / 35
350 likes | 559 Views
Retinitis Pigmentosa MR.Manaviat Yazd university of Medical sciences 2010. In the name of god. Definition :.

E N D
Retinitis PigmentosaMR.ManaviatYazd university of Medical sciences 2010
Definition: - Retinitis pigmentosa is defined as a group of hereditary disorders that diffusely involve photoreceptor and pigment epithelial function characterized by progressive visual field loss and abnormal ERG.
Definition Primary RP: The disease process is confined to the eyes with no other systemic manifestations. Secondary RP: The retinal degeneration is associated with single or multiple organ system diseases.
Ophthalmic Findings Arteriolar narrowing Waxy pallor of the disc Bone spicule-like pigment changes Atrophic appearance of peripheral retina Loss of foveal reflex Irregularity of vitreoretinal interface Cystoids macular edema Vitreous cells Posterior subcapsular cataracts
RPSymptoms : -abnormal adaptation -night blindness . - loss of midperipheral V.F -Tunel vision Signs : -Posterior subcapsular cataract . -Vitreous cells -attenuated retinal vessels . - bone spicule pigment. -waxy pallor of discs. -cystoid macular edema .
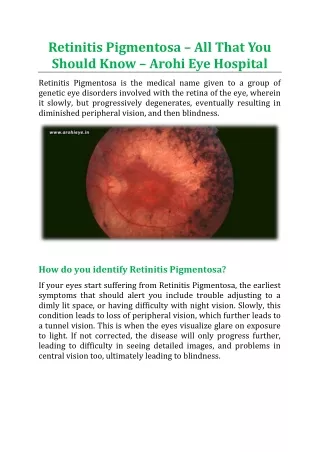
SYMPTOMS The age of onset, progression and prognosis is dependent on the mode of inheritance. In general the dominant form is of later onset and milder degree, recessive and X-linked recessive forms may present in infancy or childhood. Patients notice poor night vision (nyctalopia), visual fields become increasingly constricted and central vision may ultimately be lost.
SIGNS The three signs of typical retinitis pigmentosa are: peripheral clumps of retinal pigmentation termed (bone-spicule pigmentation) attenuation of the retinal arterioles disc pallor. Patients may also have cataracts (glare) at an early age and may develop cystoid macular oedema.
The clinical appearance of the peripheral retina in retinitis pigmentosa.
PATHOGENESIS The disease affects both types of photoreceptors but the rods are particularly affected. The inheritance may be: •autosomal recessive (sporadic cases are often in this category) •autosomal dominant •X-linked recessive. Several forms of retinitis pigmentosa have been shown to be due to mutations in the gene for rhodopsin. EPIDEMIOLOGY The prevalence of this group of diseases is 1 in 4000.
INVESTIGATION A careful family history will help to determine the mode of inheritance. The diagnosis can usually be made clinically. Electrophysiologic tests are also useful in diagnosis, particularly in early disease where there may be few clinical signs. Recent work on mapping the genetic loci for the condition has opened new avenues for genetic counselling and determining disease mechanism. The possibility of associated syndromes should be borne in mind. Usher’s syndrome, for example, is a recessive disorder characterized by deafness and retinitis pigmentosa. Retinitis pigmentosa also occurs in mitochondrial disease.
Approach to the patient with RP -History -Refraction -Perimetry -Color vision testing -Dark adaptation and ERG -Funduscopy -Genetic counseling -Psycologic counseling
Diagnosis clinical findings ERG Kinetic visual field
ERG The ERG in RP typically shows a loss or marked reduction of both rod and cone signals. Although rod loss usually predominates(reduced both a and b waves) The ERG becomes undetectable late in the course of many types of RP.
Variants of RP • Sectorial RP: involving 1 or 2 sectors of the fundus is generally symmetric in the 2 eyes. Carriers of X-Linked RP can appear as a sectorial RP. • Central RP • Pericentral RP • Unilateral RP
Special forms of RP - Some 15-20% of indivisuals with RP have associated hearing loss. (Usher syndrome) The Laurence- moon- biedel syndrome includes RP, menial retardation, polydactylism, truncal obesity and hypogonadism. (the macula is often involved early) - leber,s congenital amaurosis ia an autosomal recessive disorder associated with sever reduction in vision near birth and very reduced ERGs. - retinitis punctata albescens -progressive core- rod degeneration
Acquired causes of retinal degeneration Previous Ophthalmic artery occlusion Diffuse uveitis Infections Paraneoplastic syndromes Drug toxicity
Genetic considerations 100 genetic types 45 genes have been identified AD RP (10% - 20%) AR RP (20%) X-linked RP (10% - 25%) No family history (40%) Most common forms of RP are due to mutations in the rhodopsin gene.
Management (1) -RP is a chronic degeneration and the majority of patients do very well for decades . -Total blindness is an infrequent endpoint - Offspring are not at immediate risk of RP unless the patient has AD disease . -In usher syndrome most of the deafness is congenital . -Management of RP includes ophthalmic evaluation at intervals of 1-2 years. -Recognition of refractive errors.
Management (2) -Cataracts can be extracted if they reduce vision . -RP patients do well with intraocular lenses . -Many RP patients benefit from low vision aids. -Sunglasses
-CME may respond to oral acetazolamide. -CME that is nonresponsive to acetazolamide often shows resolution after intravitreal triamcinolone
Management (3) - high daily doses of Vitamine A Palmitate (15000 IU/day) can slow the progression of RP by 20% per year -high doses of vitamine A should not be used by pregnant women and longterm -side effect like liver toxicity must be considered. - an antioxidant mix might be helpful in RP patients. - it is likely that phototoxicity plays a role in retinal degenerations. - molecular genetics may in the future provide a means for modifying the course of RP.
- Avoidance of high dose vitamin E supplementation (400 IU/day) - in older adults longterm vitamin A has been associated with a decrease in bone denisity. - decosa hexaenoic acid (DHA) in adition to vitamin A could further slow the course of RP - DHA is thought to facilitate the release of Vitamine A in the subretinal space - vitamin A alone is though to provide seven additional years of vision, vitamin A with an oily fish diet provides almost 20 years of visual preservation for the average patient with RP.
Some future directions that may lead to treatments • Gene therapy • Neuroprotection with intravitrealciliaryneurotrophic factor • Treatment with channel blockers or drugs that interfere with apoptosis • Microchip implant • Stem cell tharapy