Download

1 / 27
350 likes | 729 Views
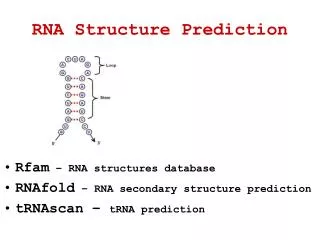
RNA Structure Prediction. Chapter 16. Primary, Secondary and Tertiary Structures. RNA Structures. Ab Initio. Prediction based on a single RNA sequence Search for RNA structure with lowest energy Free energy calculated from G-C < A-U < G-U < unpaired pairs

E N D
RNA Structure Prediction Chapter 16
Ab Initio Prediction based on a single RNA sequence Search for RNA structure with lowest energy Free energy calculated from G-C < A-U < G-U < unpaired pairs Stacking between aromatic rings (van der Waals interactions [no apostrophe]) gives rise to cooperativety Neighboring loops or bulges impose unfavorable entropic change Find all possible base-pairing interaction Calculate the energy of each and choose the lowest energy configuration Dot Matrices Plot all interactions in self alignment plot Find diagonals after applying sliding window Dynamic Programming Find the single optimal match Use Watson-Crick and wobble base pairing scores Conformations with slightly higher energies may exist without optimal base pairing
Partition Function Use a probability distribution to generate sub-optimal structures within a given energy range Mfold http://mfold.bioinfo.rpi.edu/applications/mfold/ Dynamic programming and thermodynamic calculation RNAfold http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi Extend alignment to more than one diagonal in dotplot to calculate thermodynamic stability of structures
Comparative Approach Assumption that homologous RNA sequences fold into same structure Covariation Covariant regions in homologous sequences are likely to be basepaired Predict consensus structure based onm predictions for all aligned sequences RNAalifold http://rna.tbi.univie.ac.at/cgi-bin/RNAalifold.cgi Prealignment Predictions based on covariance, minimum free eneregy, dynamioc programming finds optimal satructure for entire alignment Foldalign No prealignment http://foldalign.ku.dk/ Clustal alignment and dynamic programming
Chapter 17 Genome Mapping, Assembly and Comparison
Definitions Genomics – study of genomes Structural genomics (genome analysis) – identification of genes, annotation of gene features, comparison of genome structures Functional genomics – analysis of genome wide gene expression and gene functions
Genome Mapping • Cytological map • Banding pattern of metaphase chromosomes • Low resolution (Dustin units) • Genetic map • Relative positions of genetic markers • Marker associated with specific genetic trait • The closer the markers, the lower the probability of separation in cross-over event, and independent inheritance • Physical map • Order of clone fragments using a library of radio-labeled probes `
Genome Sequencing • Shotgun approach • Sequence large number of randomly cloned DNA fragments • Number of fragments to be sequenced is large to allow overlap to reconstruct entire genome • Requires no knowledge of physical map • Typically equivalent of 6 genome length (“6× coverage”) must be sequences to ensure correct assembly • Gaps filled in with PCR “chromosome walking” (successive sequencing from primers designed from last round of sequencing results) • Hierarchical approach • Clone of very large fragments (100-300kb) into Bacterial Artificial Chromosomes (BACs) • Map BAC inserts by restriction enzyme analysis • Arrange in order • Choose smallest number of BACs that cover entire genome (“golden tiling path”) • Sub-clone BAC insert fragments into bacterial vectors and sequence
Genome Sequence Assembly Short sequence 500bp runs → 5-10kb contigs → 30-50kb supercontigs (scaffolds) • Major challenges • Sequence errors • Vector DNA contamination (filtering programs) • Repetitive sequence regions (RepeatMasker)
Dealing with repeats (almost…) • Forward-reverse constraint
Base calling and assembly programs • Base calling:Phred • http://www.phrap.org/ • Fourier analysis to resolve fluorescent traces • Assignment to base giving probability score • Sequence assembly: Phrap • http://www.phrap.org/ • Takes Phred files as input • Performs Smith-Waterman local alignment • Progressively merge sequence pairs with highest to lowest similarity scores, removing overlaps • Outputs contigs → Nucleotide sequence
Additional software • VecScreen • To remove “contaminating” vector DNA sequences from genomes • http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html • Performs BLAST screen of submitted sequence against UniVec non-redundant vector database • Matches are displayed • TIGR Assembler (last updated 2003) • http://www.jcvi.org/cms/research/software/ • Uses forward-reverse constraints • Smith-Waterman sequence assmbly • ARACHNE • http://www.broad.mit.edu/wga/ • Gives statistical scores to overlaps • Corrects error in multiple overlaps • Outputs contigs or supercontigs • EULER • http://nbcr.sdsc.edu/euler/ • Uses shortest distance traveling salesman algorithm • Useful for assembly of sequences with repeats
Genome Annotation • Sequence • Gene structures (GenScan, FgenesH) • Predictions verified by BLAST against sequence database, cDNA and EST (GeneWise, Spidey, SIM4, EST2Genome) • Manually verified by human curators • Functional assignment of proteins by BLAST searches of protein database • Further functional description from Pfam and InterPro and literature
Gene Ontology • Uses limited vocabulary to describe • Cellular components • Biological processes • Molecular functions • Vocabulary arranged in a hierarchical manner from widest to most specific description
Automated Genome Annotation • Genome data generated at exponential rate requires automatic genome annotation • Based on homologies • Genequiz • http://swift.cmbi.kun.nl/swift/genequiz/ • BLAST and FASTA homology searches of database • Domain analysis with PROSITE and Blocks databases • Analysis of secondary and supersecondary (eg. Coiled-coils) • All results compiled to produce summary with assigned confidence level
Annotation of hypothetical proteins • In newly sequences genome as much as 40% of protein are “hypothetical” • To assign function: • Homology searches in databases • Search for similar motifs, domains and secondary structures • Identify conserved functional sites by HMM • Predict structure with fold recognition or threading • Assign broad function to protein • Test assigned function experimentally
How many genes in a genome? • Total number of human genes ~25,000 • Equivalent to that in mouse • 4× more than Saccharomyces cerevisiae • Not number of cells in organism that counts, but number of specialized cells (tissues) and response conditions
Genome Economy • One gene → one protein is not true • EST suggests >100,000 proteins in humans (from 25,000 genes?) • Alternative splicing • Joining different exons from a single transcript to form different proteins • Exon shuffling • Joining exons from different genes • Drosophila Dscam gene contains 115 exons, 20 of which are constitutively spliced and 95 of which are alternatively spliced • Expresses 38,016 different mRNAs by virtue of alternative splicing • Trans-splicing • Drosophila mdg4 gene • Joins 4 exons on sense strand and 2 exons on anti-sense strand • Single transcript of encodes dentin phosphoprotein and sialoprotein. Protein is cleaved to form two different proteins • Human transcript for Prostrate Specific Antigen (PSA) also encodes PSA-LM in 4th intron
Comparative Genomics • Compare genomes from different organisms • Whole Genome Alignment • Extent of genome conservation • Mechanism of genome evolution • MUMer and BLASTZ • Modified BLAST to align long genome sequences • Finding a minimal genome • What are the minimum number of genes to support a free-living cellular entity? • Useful to identify genes constituting essential metabolic pathways • Lateral Gene Transfer • Identify by G-C skew • GC% • Codon bias
Gene order comparisons • Where gene order is conserved between genomes, it is called synteny • Synteny may indicate functional relationships • Often indicate physical interaction of proteins • Genes encoding proteins catalyzing consecutive steps of metabolic pathway sometimes are ordered – co-regulation of “operon”? • MAL cluster in yeast: multigene complex that encodes the MAL23 trans-acting MAL-activator, MAL21 maltose permease, and MAL22 maltase in order on chromosomes 2, 3, 7, 9 and 10