Download
1 / 73
740 likes | 882 Views
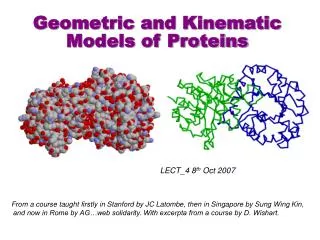
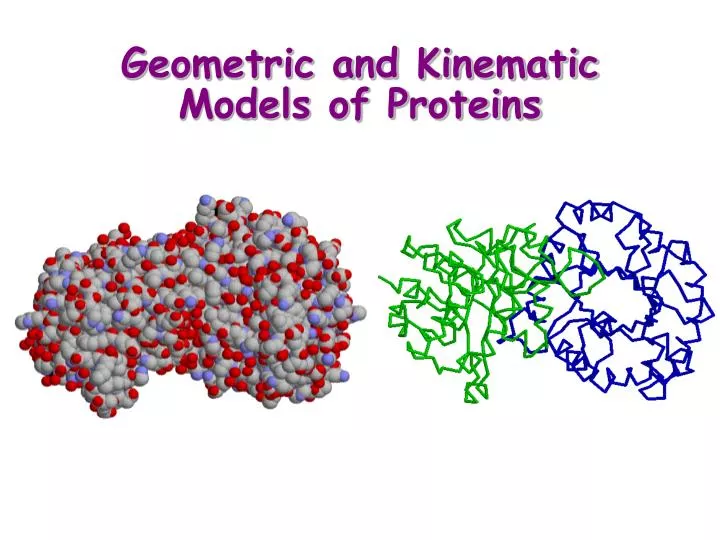
Geometric and Kinematic Models of Proteins. Study of movement independent of the forces that cause them. What is Kinematics?. Protein. Long sequence of amino-acids (dozens to thousands), also called residues from a dictionary of 20 amino-acids. Role of Geometric and Kinematic Models.

E N D
Study of movement independent of the forces that cause them What is Kinematics?
Protein • Long sequence of amino-acids (dozens to thousands), also called residues from a dictionary of 20 amino-acids
Role of Geometric and Kinematic Models • Represent the possible shapes of a protein (compare/classify shapes, find motifs) • Answer proximity queries: Which atoms are close to a given atom? (computation of energy) • Compute surface area (interaction with solvent) • Find shape features, e.g., cavities (ligand-protein interaction)
What are the issues? • Large number of atoms Combinatorial problems • Large number of degrees of freedom Large-dimensional conformation space • Need to efficiently update information during simulation (surface area, proximity among atoms): • What is the position of every atom in some given coordinate system? • Which atoms intersect a given atom? • What atoms are within some distance range from another one? • Complex metric in conformational space • Many shape matching issues
Geometric Models of Bio-Molecules • Hard-sphere model (van der Waals radii) • Van der Waals surface
Van der Waals Potential Van der Waals interactions between twoatoms result from induced polarization effect (formation of electric dipoles). Theyare weak, except at close range. The van der Waals force is the force to which the gecko's unique ability to cling to smooth surfaces is attributed! 12-6 Lennard-Jones potential
Geometric Models of Bio-Molecules • Hard-sphere model (van der Waals radii) • Van der Waals surface Van der Waals radii in Å
Geometric Models of Bio-Molecules • Hard-sphere model (van der Waals radii) • Van der Waals surface • Solvent- accessible surface • Molecular surface
Computed Molecular Surfaces Probe of 1.4Å Probe of 5Å
Computation of Hard-Sphere Surface (Grid method [Halperin and Shelton, 97]) • Each sphere intersects O(1) spheres • Computing each atom’s contribution to molecular surface takes O(1) time • Computation of molecular surface takes Θ(n) time Why?
Computation of Hard-Sphere Surface (Grid method [Halperin and Shelton, 97]) • Each sphere intersects O(1) spheres • Computing each atom’s contribution to molecular surface takes O(1) time • Computation of molecular surface takes Θ(n) time Why? D. Halperin and M.H. Overmars Spheres, molecules, and hidden surface removalComputational Geometry: Theory and Applications 11 (2), 1998, 83-102.
Trapezoidal Decomposition D. Halperin and C.R. Shelton A perturbation scheme for spherical arrangements with application to molecular modelingComputational Geometry: Theory and Applications 10 (4), 1998, 273-288.
Possible project: Design software to update surface area during molecule motion Other approach: Alpha shapes http://biogeometry.duke.edu/software/alphashapes/pubs.html
Simplified Geometric Models • United-atom model: non-polar H atoms are incorporated into the heavy atoms to which they are bonded • Lollipop model: the side-chains are approximated as single spheres with varying radii • Bead model: Each residue is modeled as a single sphere
Visualization Models • Stick (bond) model
Visualization Models • Stick (bond) model • Small-sphere model
(x4,y4,z4) (x5,y5,z5) (x6,y6,z6) (x8,y8,z8) (x7,y7,z7) (x1,y1,z1) Kinematic Models of Bio-Molecules • Atomistic model: The position of each atom is defined by its coordinates in 3-D space (x3,y3,z3) (x2,y2,z2) p atoms 3p parameters Drawback: The bond structure is not taken into account
Peptide bonds make proteins into long kinematic chains The atomistic model does not encode this kinematic structure( algorithms must maintain appropriate bond lengths)
Kinematic Models of Bio-Molecules • Atomistic model: The position of each atom is defined by its coordinates in 3-D space • Linkage model:The kinematics is defined byinternalcoordinates (bond lengths and angles, and torsional angles around bonds)
T? T? Linkage Model
Issues with Linkage Model • Update the position of each atom in world coordinate system • Determine which pairs of atoms are within some given distance(topological proximity along chain spatial proximitybut the reverse is not true)
z T(x) y T x x Rigid-Body Transform
y x 2-D Case
y y x x 2-D Case
y y x x 2-D Case
y y x x 2-D Case
y y x x 2-D Case
y y x x 2-D Case
y y Rotation matrix: cos q -sin qsin qcos q j i q ty tx x x 2-D Case
y y Rotation matrix: i1 j1i2j2 j i q ty tx x x 2-D Case
y y Rotation matrix: a i1 j1i2j2 a b j i = b’ q ty a’ b’ b q a tx a a’ x x 2-D Case v Transform of a point?
y y y’ q y ty x’cos q -sin qtxx tx + x cosq – y sin q y’ = sin q cos qtyy = ty + x sin q + y cos q 1 0 0 1 1 1 x x’ tx x x Homogeneous Coordinate Matrix i1 j1txi2 j2ty 0 0 1 • T = (t,R) • T(x) = t + Rx
? q2 q1 3-D Case
R z y x y i z j k x Homogeneous Coordinate Matrix in 3-D i1 j1 k1txi2 j2 k2tyi3 j3 k3tz 0 0 0 1 with: • i12 + i22 + i32 = 1 • i1j1 + i2j2 + i3j3 = 0 • det(R) = +1 • R-1 = RT
z y x Example cos q 0 sinq tx 0 1 0 ty -sin q 0 cos q tz 0 0 0 1 q
k q Rotation Matrix R(k,q)= kxkxvq+ cqkxkyvq- kzsqkxkzvq+ kysq kxkyvq+ kzsqkykyvq+ cqkykzvq- kxsq kxkzvq- kysqkykzvq+ kxsqkzkzvq+ cq where: • k = (kx ky kz)T • sq = sinq • cq = cosq • vq = 1-cosq
z y x y i z j k x x’ i1 j1 k1 txx y’ i2 j2 k2 tyy z’ i3 j3 k3 tzz 1 0 0 0 1 1 = Homogeneous Coordinate Matrix in 3-D (x,y,z) (x’,y’,z’) Composition of two transforms represented by matrices T1 and T2 : T2T1
Questions? What is the potential problem with homogeneous coordinate matrix?
Building a Serial Linkage Model • Rigid bodies are: • atoms (spheres), or • groups of atoms
Building a Serial Linkage Model • Build the assembly of the first 3 atoms: • Place 1st atom anywhere in space • Place 2nd atom anywhere at bond length
Building a Serial Linkage Model • Build the assembly of the first 3 atoms: • Place 1st atom anywhere in space • Place 2nd atom anywhere at bond length • Place 3rd atom anywhere at bond length with bond angle
z x y Coordinate Frame
Building a Serial Linkage Model • Build the assembly of the first 3 atoms: • Place 1st atom anywhere in space • Place 2nd atom anywhere at bond length • Place 3rd atom anywhere at bond length with bond angle • Introduce each additional atom in the sequence one at a time