Download

1 / 44
480 likes | 822 Views
PHARMACOKINETICS. DR DINESH KURIAKOSE SABAH HOSPITAL. PHARMACOKINETICS. Quantitative and mathematical analysis of drug and drug metabolite level in the body Generally applied to the process of absorption, distribution, metabolism and excretion and to their description in numerical terms

E N D
PHARMACOKINETICS DR DINESH KURIAKOSE SABAH HOSPITAL
PHARMACOKINETICS Quantitative and mathematical analysis of drug and drug metabolite level in the body Generally applied to the process of absorption, distribution, metabolism and excretion and to their description in numerical terms Study of how the body handles a drug
PHARMACOKINETIC CONSTANTS • Volume of distribution (V) • Clearance (CL) • Terminal half-life (t 1/2) They describe the behaviour of the drug in the body t ½ = K X V/CL
To determine the loading dose & the rate of infusion required to maintain a steady state concentration in plasma. • To predict the rate and extent of accumulation of the drug • To predict modification of drug dosage required in renal and hepatic disease • To determine possible effect of other agents and pathological condition on drug disposition
VOLUME OF DISTRIBUTION (V) • Apparent volume available in the body for the distribution of drug. • It represents the relation between the total amount of drug in the body and its plasma concentration. • It is not a real volume but merely a concept which helps us to understand what we observe.
V depends on: • Partition coefficient of drug • Regional blood flow • Degree of plasma protein and tissue binding
CALCULATION OF V • bucket model • Volume of water will be the volume of distribution (V) • C0 = dose/V so V = dose/C0 C0 = initial drug concentration
Drugs with V 5-20 L 1. Predominantly localized in plasma or ECF (Muscle relaxants) 2. Extensively bound by plasma proteins (Warfarin, NSAIDs) • Drugs with V 35-45 L Lipid soluble with low mol.wt. Ethyl alcohol, Urea, some Sulphonamides • Drugs with V > total body water (45 L) 1. Extensive tissue distribution of drug 2. Concentration in tissue > plasma ( Digoxin, Fentanil, Bupivacaine)
Factors affecting Vhence Initial Concentration • Patients who are dehydrated Patients who have lost blood have significantly greater plasma conc after normal dose of IV drug, increasing the likelihood of adverse effect such as hypotension • Physiological factors: In infants and pregnant women ECF is greater so water soluble drugs have greater V • Disease conditions such as Cardiac failure and Renal failure
CLEARANCE • Vol. of blood or plasma from which the drug would need to be completely removed in unit time in order to account for its elimination from the body • Rate of drug elimination (mg/min) per unit of blood or plasma conc. • [Clearance is a constant] So Rate of drug elimination is proportional to plasma conc. • CL = CLR + CLH + CLX
Renal Clearance CLR • CLR = Total drug elimination in Urine/AUC (plasma conc. Time curve during drug elimination) • CLR = CL – CLER • These values may be useful in assessing the relative importance of renal and hepatic function in drug elimination
CLR > 0.7 CL The drug is predominantly eliminated by renal clearance It will accumulate in pts with renal disease • CLR < 0.3 CL Renal failure will have little effect on drug elimination from body Hepatic metabolism and/or biliary excretion may be responsible for drug elimination
Hepatic Clearance CLH Extraction Ratio (ER) Reflects the ability of Liver to remove drugs from hepatic capillaries ( hepatic metabolism or biliary excretion) ER = (Ca – Cv) / Ca Ca = Conc in mixed portal venous and hepatic arterial blood Cv = Conc in hepatic venous blood
Elimination of drug by liver is dependent on: • Hepatic blood flow (Q) • Proportion of unbound drug in the blood • Activity of drug metabolising enzymes THUS CLH = Q X ER
CLH can also be expressed by • CLH = Q X {CLintrinsic X f /Q+ (CLintrinsic X f)} CLintrinsic - the rate at which liver cell water is cleared of drug (ml/min) i. It is independent of liver blood flow ii. Represents the maximum ability of liver to irreversibly eliminate the drug by metabolism and/or biliary excretion f=fraction of unbound drug in the blood
CLintrinsic is relatively low than LBF • CLH = Q X { CLintrinsicXf / Q+(CLintrinsicXf)} CLH = Q X { CLintrinsicXf/Q} = CLintrinsic X f This type of clearance is known as Capacity limited or Restrictive elimination. Phenytoin, Theophylline, Warfarin, most Barbiturates and Benzodiazepines
Restrictive Elimination The drugs cleared by restrictive elimination have • Limited first pass effect after oral administration • Low hepatic extraction ration < 0.3 • Clearance is low and unaffected by liver blood flow • Clearance is profoundly influenced by changes in hepatic enzyme activity (caused by drugs, malnutrition, age, liver disease)
CLH = CLintrinsic X f • Capacity limited or restrictive elimination is also dependent on fraction of unbound drug in the blood. Capacity limited Binding insensitive Drugs that are less protein bound (20-30%) Clearance, elimination and t1/2 are less affected by changes in protein binding Capacity limited Binding sensitive Drugs are extensively protein bound Clearance and elimination are affected by changes in protein binding
CLintrinsic > LBF CLH = Q X {(CLintrinsic X f)/Q+(CLintrinsic X f)} As Q is low, Q + (CLintrinsic X f) = CLintrinsic X f Hence CLH = Q This is Flow limited or Non-restrictive elimination B antagonist, Opioid analgesics, Lidocaine
Drugs with Flow Limited Clearance • High Hepatic extraction ratio • Substantial first pass effect after oral admn • Half-life modified by changes in LBF • Insensitive to changes in hepatic enzyme activity and plasma protein binding
Practical Consideration of Pharmacokinetic Constants • To calculate the oral or IM dose from CL if desired plasma conc. is known Req. Dose = (Cp X I X CL)/F • To calculate loading and infusion dose in order to produce accurate and constant plasma conc. during infusions Loading dose=Cp X V Infusion dose=Cp X CL • BET method for calculating infusion regimes to produce constant & reproducible plasma conc. Use compartmental models • To predict the effects of altered hepatic and renal function on plasma conc. of drug
PLASMA HALF-LIFE Defined as time required for plasma concentration of drug to decrease by 50% during terminal phase of decline. t ½ = K×V/CL where K is elimination rate constant. cont….
It often reflects the duration of drug action. eg. Muscle relaxants. • This relation may not be present with other drugs such as • Opioids which crosses BBB to produce the effect. • Drugs which act irreversibly. (ChE inhibitors, MAOI). • Drugs whose action is terminated by redistribution. • Drugs with active metabolites like morphine and diazepam.
CONTEXT SENSITIVE HALF-LIFE OR TIME • Time required for plasma concentration to decrease by 50% at the end of the period of infusion designed to maintain a constant concentration. • Amount of drug accumulating in the body increases with the duration of the infusion. • Provide a useful practical indication of the decline in plasma concentration after infusion of defined duration. • Provides a guide likely duration of drug activity and speed of recovery after infusion of different duration.
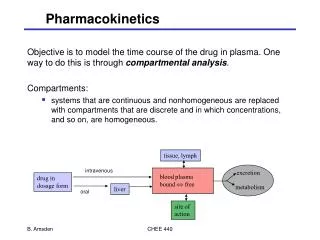
MEASUREMENT OF V,CL,T ½ BY COMPARTMENTAL ANALYSIS. • V and CL of drugs can be determined from the decline in plasma concentration by compartmental or non-compartmental methods. • In compartmental methods suitable models are used to describe the distribution and elimination of drugs. • Values of V and CL are derived from the parameters of the model.
One Compartmental model V= dose/Co CL=dose/AUC
Plasma Conc. – Time Curve α = In2/t1/2 α Β = In2/t1/2 β Cp = A. e-αt +B. e-βt
Two Compartment Open Model Cp = A. e-αt +B. e-βt AUC=A/α+B/β CL=dose/AUC K21=Aβ+Bα/A+B K10 =αββ/K21 K12 =α+β-(K21+ K10)
Varea = dose/β(AUC) Vss =[dose/A+B] × [K12 +K21/K21]
NON-COMPARTMENTAL METHODS • Problems associated with resolution of plasma concentration data by specific compartmental models has led to use of non-compartmental methods of pharmacokinetic analysis.
CL=dose/AUC AUC is area under plasma concentration-time curve between t=0 and t=∞. AUC can be estimated by trapezoidal route Terminal area=final plasma conc. X t1/2/0.693 MRT = AUMC/AUC MRT – Mean Residence Time AUMC-total area under the 1st moment of the plasma conc time curve Vss =CL×MRT
NON-LINEAR OR SATURATION KINETICS • Physiological processes concerned with drug distribution and elimination are dependent on carrier transport. They are saturable, i.e. finite transport capacity. • Reaction concerned with metabolism are saturable and proceed at constant rate at high substrate conc. • In this situation drug transport or drug metabolism occurs at the constant rate independent of drug conc. This is saturation or non-linear kinetics. • dx/dt=-K (Zero order process)
MICHAELIS MENTEN KINETICS • Saturation of metabolic reactions results in change of metabolism from first order process dx/dt = -Kx to zero order process dx/dt=-k and is constant and independent of drug conc. V=Vmax × Cp/ Km+Cp V-rate of drug elimination Vmax-max. rate of drug elimination Km-affinity constant
When Cp < Km V ∞ Cp (first order process) When Cp > Km V ∞ Vmax./Km (constant) zero order process • In zero order kinetics, all kinetics parameter are dependant of dose of drug. • In first order kinetics, all kinetics parameter are independent of dose of drug. • Saturation kinetics may occur during elimination of large and repeated dose of thiopentone.
BIO-AVAILABILITY • Rate and extent to which a drug is absorbed and becomes available at the site of action. • Proportion of dose that is present in systemic circulation • Absolute bioavailability % = AUC0-α after oral administration / AUC0-α after IV administration
DRUGS WITH HIGH BIO-AVIALABILITY • Stable in GI secretion • Well absorbed from small intestine • Not significantly metabolised by the gut wall or liver before they gain access to systemic circulation eg. Digoxin, diazepam, warfarin and phenytoin
DRUGS WITH LOW BIO-AVAILABILITY • Broken down in GI tract. Benzylpencillin / heparin • Absorbed to limited extent because of physical chemical factors such as lipid solubility and molecular weight (neostigmine and glycopyrrolate) • High first pass effect extensive metabolism by gut wall and liver. Morphine, propranolol, lidocaaine.