Download

1 / 60
620 likes | 1.33k Views
Pharmacokinetics. ผศ.มนุพัศ โลหิตนาวี manupatl@nu.ac.th manupatl@hotmail.com. Outline. Introduction Physicochemical properties Absorption, Bioavialability, routes of admistration Distribution Biotransformation (Metabolism) Excretion Clinical pharmacokinetics.

E N D
Pharmacokinetics ผศ.มนุพัศ โลหิตนาวี manupatl@nu.ac.th manupatl@hotmail.com
Outline • Introduction • Physicochemical properties • Absorption, Bioavialability, routes of admistration • Distribution • Biotransformation (Metabolism) • Excretion • Clinical pharmacokinetics
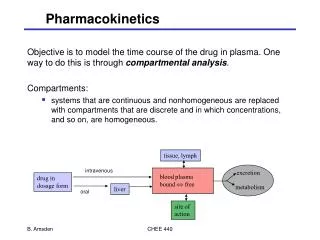
Components of pharmacokinetics • Input, dosing by using routes of administration • Pharmacokinetic processes (figure 1, drawing) • Absorption • Distribution • Biotransformation (Metabolism) • Excretion
Cell membrane • barrier of drug permeation (drawing), with semipermeable property • factors affecting drug across cell membrane • cell membrane properties • physicochemical properties of drugs
Cell membrane • physicochemical properties of drugs • size and shape • solubility • degree of ionization • lipid solubility
Cell membrane • Characteristics of Cell membrane • Lipid bilayer: mobile horizontally, flexible, high electrical resistance and impermeable to high polar compounds • protein molecules function as receptors or ion channels or sites of drug actions.
Diffusion across the cell membrane • Passive transport (drawing) • higher conc to lower conc area • energy independent • at steady state both sides have equal conc.(non electrolye cpds) • electrolyte: conc. of each side depends on pH (fig 2) • weak acid and weak base
Diffusion across the cell membrane • Carrier-mediated membrane transport (drawing) • lower conc to higher concentration area (agianst concentration gradient) • structure specific • rapid rate of diffusion • Active and Facillitated transport
Diffusion across the cell membrane • Active transport • energy dependent • structure specific, inhibited by structure-related cpds, saturable • Facillitated transport • energy independent • structure specific, inhibited by structure-related cpds, saturable
Saturable process • Drawing • almost all protein-mediated process in our body can occur this process saturation not only transport system but also others such as enzymatic reaction, drug-ligand binding and so on. • because functional protein molecules are limited.
Drug absorption • Parameters in drug absorption • Rate constant of drug absorption (Ka) • Bioavialability (F) • Anatomical aspects affecting absorption parameters (Drawing) • GI tract (metabolzing organ and barrier of drug movement) • Liver (portal and hepatic vien, excretion via biliary excretion) • cumulative degradation so called “First pass effect”
Drug absorption • Factors affecting drug absorption (Drawing) • Physicochemical properties of drugs • pH at site of absorption • Concentration at the site of administration • Anatomical and physiological factors • Blood flow • Surface area
Routes of administration • Enteral and parenteral routes • Pros and cons between Enteral and parenteral
Enteral administration • Pros • most economical, • most convenient • Cons • high polar cpds could not be absorbed • GI irritating agents • enzymatic degradaion or pH effect • Food or drug interaction (concomitant used) • cooperation of the patients is needed • first pass effect due to GI mucosa
Parenteral administration • Pros • Rapidly attained concentration • Predictable conc by the calculable dose • Urgent situation • Cons • Aseptic technic must be employed • Pain • limited self adminstration • More expensive
Enteral administration • Common use of enteral administration • Oral administration • Sublingual administration • Rectal administration
Enteral administration • Concentrion-time course of oral administration (Drawing) • Rapid increase in plasma conc until reaching highest conc and subsequent decrease in plasma conc • Drawing (concept of MTC and MEC) • Absorption phase • Elimination phase
Enteral administration • Prompt release: the most common dosage form • Special preparation: Enteric-coat, SR • SR, Controlled release: Purposes and limitation
Enteral administration • Sublingual administration • Buccal absorption • Superior vana cava directly: no first pass effect • Nitroglycerin (NTG): highly extracted by the liver, high lipid solubility and high potency (little amount of absorbed molecules be able to show its pharmacological effects and relieve chest pain).
Enteral administration • Rectal adminstration • unconscious patients, pediatric patients • 50% pass through the liver and 50% bypass to the inferior vena cava • lower first pass effect than oral ingestion • inconsistency of absorption pattern • incomplete absorption • Irritating cpds
Parenteral administration • Common use of parenteral administration • Intravenous • Subcutaneous • Intramuscular • Simple diffusion • Rate depends on surface of the capillary, solubility in interstitial fluid • High MW: Lymphatic pathway
Parenteral administration • Intravenous • precise dose and dosing interval • No absorption (F=1), all molecules reach blood circulation • Pros: Calculable, promptly reach desired conc., Irritating cpds have less effects than other routes • Cons: unretreatable, toxic conc, lipid solvent cannot be given by this route (hemolysis), closely monitored
Parenteral administration • Subcutaneous • suitable for non-irritating cpds • Rate is usually slow and constant causing prolonged pharmacological actions.
Parenteral administration • Intramuscular • more rapid than subcutaneous • rate depends on blood supply to the site of injection • rate can be increased by increasing blood flow (example)
Pulmonary absorption • gaseous or volatile substances and aerosol can reach the absorptive site of the lung. • Highly available area of absorption • Pros: rapid, no first pass effect, directly reach desired site of action (asthma, COPD) • Cons: dose adjustment, complicated method of admin, irritating cpds.
Bioequivalence • Pharmaceutical equivalence (drawing) • Bioequivalence: PharEqui+ rate+ bioavialable drugs • Factors: • Physical property of the active ingredient: crystal form, particle size • Additive in theformulation: disintegrants • Procedure in drug production: force
An example of a generic product that could pass a bioequivalence test: Simvastatin (Parent form, n=18)
An example of a generic product that could pass a bioequivalence test: Ondansetron (n=14)
An example of a generic product that could pass a bioequivalence test: Clarithromycin (n=24)
Distribution • Drawing • distribution site: well-perfused organs, poor-perfused organs, plasma proteins • Well-perfused: heart, liver, kidney, brain • Poor-perfused: muscle, visceral organs, skin, fat
Distribution • Plasma proteins • Albumin: Weak acids • alpha-acid glycoprotein: Weak bases • Effects of plasma protein binding • Free fraction: active, excreted, metabolized • the more binding, the less active drug • the more binding, the less excreted and metabolized: “longer half-life”
Distribution • Effects of well distribution into the tissues • deep tissue as a drug reservoir • sustain released drug from the reservoir and redistributed to the site of its action • prolong pharmacologic actions
Distribution CNS and CSF • Blood-Brain Barrier (BBB) • unique anatomical pattern of the vessels supplying the brain • only highly lipid soluble compounds can move across to the brain • infection of the meninges or brain: higher permeability of penicillins to the brain.
Distribution Placental transfer • Simple diffusion • Lipid soluble drug, non-ionized species • first 3 mo. of pregnancy is very critical: “Organogensis”
Biotransformation • Why biotranformed? (Figure 5) • Normally, drugs have high lipid solubility therefore they will be reabsorbed when the filtrate reaching renal tubule by using tubular reabsorption process of the kidney. • Biotransformation changes the parent drug to metabolites which always have less lipid solubility (more hydrophilicity) property therefore they could be excreted from the body
Biotransformation • Biotransformation • to more polar cpds • to less active cpds • could be more potent (M-6-G) or more toxic (methanol to formaldehyde)
Biotransformation • Phase I and II Biotransformation • Phase I : Functionization, Functional group • Phase II: Biosynthetic, Molecule
Biotransformation • Phase I Reactions (Table 2) • Oxidation • Reduction • Hydrolysis
Biotransformation • Phase II Reactions (Table 3) • Glucuronidation • Acetylation • Gluthathione conjugation • Sulfate conjugation • Methylation
Biotransformation • Metabolite from conjugation reaction • Possibly excreted into bile acid to GI • Normal flora could metabolize the conjugate to the parent form and subsequently reabsorbed into the blood circulation. This pheonomenon is so-called “Enterohepatic circulation” which can prolong drug half-life.
Biotransformation • Site of biotransformation • Mostly taken place in the liver • Other drug metabolizing organs: kidney, GI, skin, lung • Hepatocyte (Drawing)
Biotransformation The Liver:Site of biotransformation: • mostly enzymatic reaction by using the endoplasmic reticulum-dwelling enzymes.(Phase I), Cytosolic enzymes are mostly involved in the phase II Rxm. • Method of study phase I Rxm • Breaking liver cells • Centrifugation very rapidly • microsomes and microsomal enzymes
Biotransformation • Cytochrome P450 monooxygenase system (figure 6) • microsomal enzymes • Oxidation reaction using reducing agent (NADPH), O2 • System requirement • Flavoprotein (NADPH-cytochrome P450 reductase, FMN+FAD) fuctions as an electron donor to cytochrome c. • Cytochrome P450 (CYP450)
Biotransformation • Steps in oxidative reactions (figure 6) • Step 1:Parent + CYP450 • Step 2:Complex accepts electron from the oxidized flavoprotein • Step 3:Donored electron and oxygen forming a complex • Step 4: H2O and Metabolite formation
Biotransformation • CYP450 is a superfamily enzyme, many forms of them have been discovered (12 families). • Important CYP450 families in drug metabolism (Fig. 7) • CYP1 (1A2) • CYP2 (2E1, 2C, 2D6) • CYP3
Biotransformation • Factors affecting biotransformation • concurrent use of drugs: Induction and inhibition • genetic polymorphism • pollutant exposure from environment or industry • pathological status • age
Biotransformation • Enzyme induction • Drugs, industrial or environmental pollutants • increase metabolic rate of certain drugs leading to faster elimination of that drugs. • “autoinduction” • Table 4
Biotransformation • Enzyme induction • important inducers: • antiepileptic agents, glucocorticoids for CYP3A4 • isoniazid, acetone, chronic use of alcohol for CYP2E1
Biotransformation • Enzyme inhibition:(drawing) • Competitive binding and reversible: Cimetidine, ketoconazole, macrolide metabolites • Suicidal inactivators: Secobarbital, norethindrone, ethinyl estradiol • Clinical significance: erythormycin and terfenadine or astemizole causing cardiac arrhythmia.
Biotransformation • Genetic polymorphism • Gene directs cellular functions through its products, protein. • Almost all enzymes are proteins so they have been directed by gene as well. • Drug-metabolizing enzymes: • Isoniazid: causing more neuropathy in caucaasians leading to identification of the first characterized pharmacogenetics. • due to the rate of N-acetylation: Slow and fast acetylators