Download

1 / 46
510 likes | 799 Views
Idiopathic Pulmonary Fibrosis. OBJECTIVES. Know the definitions of ILD, IIP, and IPF. Understand the pathogenesis of IPF. Appreciate the clinical features. Realize how the diagnosis of IPF is made. Know current therapies.

E N D
OBJECTIVES • Know the definitions of ILD, IIP, and IPF • Understand the pathogenesis of IPF • Appreciate the clinical features • Realize how the diagnosis of IPF is made • Know current therapies • Become aware of areas of current research and novel therapeutic approaches • Be able to summarize current thinking about IPF
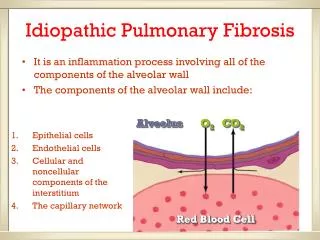
Interstitial Lung Disease (ILD) or Diffuse Parenchymal Lung Disease (DPLD) • Any process that results in inflammatory-fibrotic infiltration of the alveolar septa resulting in effects on the capillary endothelium and alveolar epithelium. • Generic term used to describe many conditions that cause breathlessness and/or cough and are associated with radiographic bilateral lung abnormalities. Mason: Murray & Nadel's Textbook of Respiratory Medicine, 4th ed. • “Death occurred about three months and a half after the onset of the acute disease and the lung was two thirds of the normal size, grayish in color, and hard as cartilage. Microscopically these areas showed advanced fibrotic changes and great thickening of the alveolar walls.” • - Sir William Osler, 1892
INTERSTIAL LUNG DISEASES Connective Tissue Diseases Scleroderma Polymyositis-Dermatomyositis Systemic Lupus Erythematosus Rheumatoid Arthritis Mixed Connective Tissue Disease Ankylosing Spondyitis Treatment-Related / Drug-Induced Antibiotics – nitrofurantoin, sulfasalazine Antiarrhythmics – amiodarone, propanolol Anti-inflammatories – gold, penacillamine Anti-convulsants – dilantin Chemotherapeutic agents – bleomycin, cyclophosphamide, methotrexate, azathioprine Therapeutic radiation Oxygen toxicity Narcotics Primary (Unclassified) Sarcoidosis Langerhans cell histiocytosis Amyloidosis Pulmonary vasculitis Lipoid pneumonia Lymphangitic carcinomatosis Bronchoalveolar carcinoma Pulmonary lymphoma Gaucher’s Disease Niemann-Pick Disease Hermansky-Pudlak syndrome Neurofibromatosis Lymphangioleiomyomatosis Tuberous Sclerosis ARDS AIDS Bone Marrow Transplantation Postinfectious Eosinophilic pneumonia Alveolar Proteinosis Diffuse Alveolar Hemorrhage Syndromes Alveolar microlithiasis Metastatic calcification Occupational and Environmental Diseases Inorganic Organic Silicosis Bird breeder’s lung Asbestosis Farmer’s lung Hard-metal pneumoconiosis Bacteria – e.g. NTB mycobacteria Coal worker’s pneumoconiosis Fungi – e.g. Aspergillus Berylliosis Animal protein – e.g. Avian Aluminum oxide fibrosis Chemical sensitizers - Talc pneumoconiosis e.g. isocyanates Siderosis (arc welder) Stannosis (tin) Idiopathic Fibrotic Disorders Acute interstitial pneumonitis (Hamman-Rich syndrome) Idiopathic Pulmonary Fibrosis Familial Idiopathic Pulmonary Fibrosis Desquamative intersitial pneumonitis Respiratory bronchiolitis Cryptogenic organizing pneumonia Nonspecific interstitial pneumonitis Lymphocytic interstitial pneumonia (Sjögrens Syndrome, AIDS, Hashimoto’s) Autoimmune pulmonary fibrosis (inflammatory bowel disease, PBC, ITP, AIHA)
ATS/ERS International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias, Am J Respir Crit Care Med. 2002
QUICK HISTORY OF IIP • In 1969, Liebow and Carrington described 5 types of chronic interstitial pneumonias based on histology: • Usual interstitial pneumonia (UIP) • Bronchiolitis obliterans interstitial pneumonia and diffuse alveolar damage (BIP) • Desquamative interstitial pneumonia (DIP) • Lymphocytic interstitial pneumonia (LIP) • Giant cell interstitial pneumonia (GIP) • In 2002, the ATS/ERS published their consensus classification of IIP based on Clinical-Radiologic-Pathologic categories:
ATS/ERS International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias, Am J Respir Crit Care Med.
USUAL INTERSTITIAL PNEUMONIA PATTERN • The UIP pattern can be seen in the following conditions: • IPF • Familial IPF • Collagen vascular diseases • Drug toxicity • Chronic hypersensitivity pneumonitis • Asbestosis • Hermansky-Pudlak syndrome The term UIP is usually reserved for patients in whom the lesion is idiopathic UIP ≈ IPF
USUAL INTERSTITIAL PNEUMONIA PATTERN Key histologic features: • Dense fibrosis with remodeling of lung architecture , frequent honeycomb fibrosis • Fibroblastic foci usually at the edge of scarring • Patchy lung involvement • Usually subpleural distribution Important negative findings: • No active lesions typical of other ILD’s • Lackof marked interstitial chronic inflammation • No (or rare) granulomas • No evidence of inorganic dust deposits (e.g. asbestos bodies) • Lack of marked eosinophilia
USUAL INTERSTITIAL PNEUMONIA PATTERN Mason: Murray & Nadel's Textbook of Respiratory Medicine, 4th ed. Idiopathic Pulmonary Fibrosis, Gross and Huninghake, NEJM, 2001.
HONEYCOMB PATTERN Pictures taken from http://mediswww.meds.cwru.edu/ecsample/yeartwo/pulmonary/interstitial.html
IDIOPATHIC PULMONARY FIBROSIS ATS definition: “IPF is a distinctive type of chronic fibrosing interstitial pneumonia of unknown cause limited to the lungs and associated with a surgical lung biopsy showing a histologic pattern of UIP.” • A distinct type of chronic fibrosing interstitial pneumonia • Unknown cause • Limited to the lungs • Associated with a histologic pattern of usual interstitial pneumonia (UIP)
EPIDEMIOLOGY • Estimated to affect approx 5 million people worldwide • The most common (and deadly) interstitial lung disease • Most cases are sporadic, but rare cases of familial IPF have been described Raghu et. al., Am J of Resp Crit Care Med 2006
EPIDEMIOLOGY Raghu et. al., Am J of Resp Crit Care Med 2006
CLINICAL PRESENTATION • Middle age 50-70s • New onset of progressive exertional dyspnea and non-productive cough • Most have symptoms for 12-18 months prior to definitive evaluation • Constitutional symptoms are uncommon • Weight loss, fever, fatigue, myalgias, or arthralgias occasionally present • Detailed occupational and exposure history
PHYSICAL EXAM • Bibasilar late inspiratory fine crackles (Velcro rales) • Tachypnea • Clubbing – 40-75% - late in disease course • Cardiac exam usually normal until middle-late stages • - augmented P2, right-sided heave, S3 gallop • Cyanosis • Rash, arthritis, myositis should suggest an alternate diagnosis
CXR 16% of patients with ILD have normal chest x-rays Idiopathic Pulmonary Fibrosis, Gross and Hunninghake, NEJM, 2001. Courtesy of W. Richard Webb, MD.
CXR Mason: Murray & Nadel's Textbook of Respiratory Medicine, 4th ed.
PFTs PFT’s = Restrictive pattern Reduced TLC, VC, and/or RV (decreased compliance) Normal or increased FEV1/FVC Decreased DLCO Source: images.md
ABG ABG = Hypoxemia, respiratory alkalosis Decreased PaO2 with rest or exercise Increased A-a gradient Other lab tests that might be useful? Elevated ESR Hypergammaglobulinemia Low-titer positive ANA (21% patients with IPF) RF Circulating immune complexes Cryoimmunoglobulins
HIGH RES CT • Can be used to detect disease, especially in pts with no or minimal changes on CXR • Can determine extent and severity of disease activity • Can now be used to differentiate IPF from other ILD Peripheral, subpleural fibrosis Alternating areas of normal tissue Honeycombing Traction bronchiectasis Later stages - more diffuse reticular pattern prominent in lower lung zones associated with thickened interlobular septa Idiopathic Pulmonary Fibrosis, Gross and Hunninghake, NEJM, 2001.
BAL in IPF • Role and value of serial BAL in IPF previously unknown • Increased inflammatory cells in IPF, but no predominant type • Kinder et al, Chest, Jan 2008 156 subjects with biopsy proven UIP/IPF enrolled between 1982-1996 BAL within 3 weeks of lung biopsy Linear relationship betweenincreasing neutrophil percentage and the risk of mortality Each doubling in the neutrophil percentagewas associated with a nearly 30% increased risk of death ortransplantation in adjusted analysis ([HR] 1.28; 95% CI, 1.01 to 1.62; p = 0.04). There was no association with lymphocyte or eosinophil percentage. Suggests that BAL fluid neutrophilpercentage at the time of diagnosis of IPF is an independentpredictor of time to death.
LUNG BIOPSY • Gold Standard for diagnosis of IPF (and IIP’s) • Large piece of lung parenchyma is required, optimally from several sites • Transbronchial biopsy is only useful for ruling out other disorders • Can be performed by thoracotomy, thorascopy, or VATS OTHER STUDIES IN IPF • Gallium Scanning (67Ga) used for staging “alveolitis” in ILD, e.g sarcoidosis • Not useful – difficult to interpret, very low specificity • VQ scan reveals patchy, non-segmental areas of decreased V • decreased perfusion in lower lung zones • increased perfusion of upper lung zones (due to PH)
ATS/ERS CRITERIA FOR DIAGNOSIS OF IPF IN ABSENCE OF SURGICAL LUNG BIOPSY Major Criteria: Exclusion of other known causes of ILD Abnormal PFTs that include evidence of restriction and impaired gas exchange Bibasilar reticular abnormalities with minimal ground glass opacities on HRCT Transbronchial lung biopsy or BAL showing no features to support alternative dx Minor Criteria: Age > 50 Insidious onset of otherwise unexplained dyspnea on exertion Duration of illness greater than 3 months Bibasilar inspiratory crackles (dry or “Velcro”-type in quality) ALL of the major criteria plus at least THREE minor criteria.
NATURAL HISTORY / PROGNOSIS • Worst prognosis of all the ILD’s • Disease course is variable • 5-year survival rate is 30-50% • Median survival after diagnosis is less than 3 years • 40% IPF patients die of respiratory failure • Others die of complicating illnesses, mainly CAD and infections • End-stage disease is characterized by severe PH with cor pulmonale that does not improve with oxygen • Incidence of bronchogenic carcinoma is increased in patients with IPF • Factors associated with shortened survival: • Increased neutrophil count • older age • poor pulmonary function at presentation • recent deterioration in results of PFT’s • advanced fibrosis
ACUTE EXACERBATIONS OF IPF • Traditional view: slow, steady decline in lung fuction à respiratory failure • Several recent clinical trials have shown that multiple subclinical and • acute exacerbations lead to decline in pulmonary function • Martinez et al, Ann Intern Medicine, 2005 168 patients in the placebo group of a trial evaluating interferon-γ1b (mild-mod IPF) Measures of physiology and dyspnea assessed at 12-week intervals; hospitalizations; and the pace of deterioration and cause of death over a median period of 76 weeks. Minimal physiologic deterioration or worsening severity of dyspnea over time Frequent hospitalizations for respiratory disorders (23%, 21% died) IPF was the primary cause of death in 89% who died Acute clinical deterioration preceded death in 47% • Kim et al, Eur Resp J, 2006 Analysis of 147 IPF patients demonstrated 2-year frequency of acute exacerbations was 9.6%
ACUTE EXACERBATIONS OF IPF Consensus group in 2007 defined acute exacerbation: • Diagnosis of IPF • Unexplained development of worsening of dyspnea within 30 days • HRCT with new ground-glass abnormalities • No evidence of pulmonary infection by ET aspirate or BAL • Exclusion of alternative causes, e.g. HF, PE Treatment: • Broad-spectrum antibiotics • High-dose steroids (prednisone 1 mg/kg)
GENETIC SUSCEPTIBILITY? Up to 3% of cases of IPF appear to cluster in families (Familial IPF) Armanios et al, NEJM 2007. • 73 probands from the Vanderbilt Familial Pulmonary Fibrosis Registry formutations in hTERT and hTR (telomerase RT and telomerase RNA) • Demonstrated that mutation was inherited in autosomal dominant fashion with variable penetrance • Those with IPF had mutant telomerase and short telomeres • Telomeres shorten with each cell division and ultimately lead to apoptosis • Proposed that fibrosis occurs due to death of alveolar cells
ASSOCIATED RISK FACTORS • Up to 75% of index patients with IPF are current or former smokers • Latent viral infections have also been reported to have an association • Given the similarity between asbestosis and IPF, is there a causative environmental agent? • Chronic aspiration?
GERD AND IPF Raghu et al, Eur Resp J, Oct 2006. • 65 consecutive patients with IPF were subjected to 24-h pH monitoring and esophageal manometry • 133 patients with intractable asthma and GERD used for comparison • Prevalence of GERD in IPF patients was 87% but only 47% had symptoms • GERD was higher in IPF patients (76% versus 57%; p = 0.020) • Despite tx with standard dose PPI, 12/19 still had abnormal pH • Conclusion: GERD is highly prevalent and often clinically occult in patients with IPF, and often does not respond entirely to standard dose PPI
PATHOGENESIS Originally thought inflammation à fibrosis Animal models Early IPF is dominated by inflammatory cells Asymptomatic relatives of patients with familial IPF have evidence of alveolitis in the absence of disease Alveolar macrophage thought to play a major role Secretes proinflammatory and profibrotic cytokines Promote collagen deposition PROBLEMS: 1) Little inflammation is seen histologically 2) Measurements of inflammation do not correlate 3) Anti-inflammatory therapies DO NOT WORK!
PATHOGENESIS • Starting around 1998, studies began to demonstrate that inflammation is NOT a prominent finding in most cases of IPF/UIP. • These sites are typical in alveolar epithelial injury • Abnormal wound healing involving epithelial cells and fibroblasts • Activated epithelial cells release potent fibrogenic molecules and cytokines, such as TNFα and TGFβ1
TREATMENT ATS recommendation (2000): Prednisone + Azathioprine or Cyclophosphamide Consensus recommendation (2008): Prednisone + Azathioprine + N-acetylcysteine
STEROIDS Cochrane Systematic Review in 2003 Fifteen studies were selected as potentially eligible for meta-analysis. After further analysis of full text papers, no RCTs or CCTs were identified as suitable and therefore no data was available for inclusion in any meta-analysis. All studies were excluded due to inadequate methodologies. • Currently there is no evidence to support the routine use of corticosteroids alone in the management of IPF.
1.0 0.8 Azathioprine + Prednisone (n = 14) Probability of Survival 0.6 0.4 Prednisone (n = 13) 0.2 0 0 1 2 3 4 5 6 7 8 9 Years AZATHIOPRINE • Raghu et al, Am Rev Respir Dis 1991. • 27 newly diagnosed patients with IPF • Prednisone + Azathioprine vs. Prednisone + Placebo, follow-up 9 years • After 1 year, P+A had better lung function, but was not significant • 43% (6/14) died vs. 77% (10/13) P = 0.16 P = 0.02 (age adjusted) • Side effects: leukopenia, • GI-related Raghu G, et al. Am Rev Respir Dis. 1991;144:291-296.
CYCLOPHOSPHAMIDE • Collard et al, Chest, 2004 • Retrospective study looking at 164 patients with IPF from 1984-2002 • 82 patients on prednisone and oral cyclophosphamide vs. 82 patients on prednisone alone • No difference in survival from time of initial visit • Multiple other small studies have been unimpressive • Toxicity is major (pancytopenia, hemorrhagic cystitis, GI-related)
Mortality, P = NS 7/80 (9%) NAC+Pred+Aza Pred+Aza+ Placebo 8/75 (11%) N-acetylcysteine (NAC) • Demedts et al, NEJM, 2005. • 182 patients with UIP • Prednisone + Azathioprine + High-dose NAC (600mg TID) vs. P/A • Significant difference in the deterioration of VC and DLCO at 12 months • Relative difference of 9% and 24% respectively • Oxidant-antioxidant imbalance?
LUNG TRANSPLANT • IPF is the most common ILD among referrals for transplant and the 2nd most frequent disease for which lung transplantation is performed • Criteria: Evidence of UIP plus any of the following: • DLCO < 39% predicted • Decrement in FVC > 10% during 6 months • Decrease in pulse ox below 88% during 6-minute walk test • Honeycombing on HRCT • 5-year survival for lung transplant in IPF is 40-50% • SLT has been the standard treatment • Living donor lobar lung transplant (LDLLT) - Date et al, Chest 2005 • 9 patients with IIP dependent on systemic steroids (up to 50mg/day) • Transplant of two lower lobes from two healthy relatives • After 10-48 months of follow-up 8/9 still alive (one died of acute rejection)
PERFENIDONE Mechanism of Action: • inhibits TGF-β-stimulated collagen synthesis • decreases the extracellular matrix • blocks fibroblast proliferation in-vivo Currently in Phase III trials in the U.S. (CAPACITY) Phase III trial in Japan ended last month: • Included 267 patients in 73 different centers • Pirfenidone 1800 mg/day vs. 1200 mg/day vs. placebo • VC, SpO2 on exertion, number of acute exacerbations were primary endpoints • At week 52: Difference in VC between groups was 70mL and 80mL • No significant difference in lowest SaO2 on exertion • No significant difference in the number of acute exacerbations • Significant difference in progression-free survival Adverse effects: rash, GI effects, fatigue
OTHER TREATMENT OPTIONS Interferon gamma-1b: • important in “wound healing” • PCRT suggested a possible mortality benefit • Large multinational trial (INSPIRE) was stopped when the primary endpoint of mortality benefit was not achieved Limited data: • Methotrexate • Cyclosporine • Colchicine • Penicillamine
SUMMARY • IPF is the most common ILD with the worst prognosis • The most important distinction is differentiate IPF from the other IIP’s • Biopsy is the gold standard for diagnosis, histology = UIP pattern with fibroblast foci (hallmark of IPF) • Most common presentation is 50-60 y.o. male with progressive dyspnea and non-productive cough • Most common physical exam findings are “Velcro” rales +/- clubbing • Most important diagnostic studies are CXR, PFT’s, ABG, and HRCT • Higher BAL neutrophil percentage at time of diagnosis = worse prognosis? • If certain clinical criteria are met, can diagnose IPF without biopsy • Acute exacerbations are now recognized to be an important target for therapy
SUMMARY • Possible genetic component involving mutant telomeres, resulting in apoptosis of alveolar cells • Newly accepted hypothesis that fibrosis is a result of aberrant “wound healing” resulting from repeated injury of unknown cause • There is a high correlation with GERD in IPF • There is still no effective therapy for IPF • Current recommendation is steroids + azathioprine + NAC • SLT improves 5-year survival, LDLLT shows promise in advanced disease • Perfenidone will likely be the next option in therapy for IPF • There are a number of novel therapies on the horizon