Download

1 / 30
330 likes | 936 Views
JA Carde, PhD Modificado de Dra. Liza Jiménez UPR-Ag Lab Biol 4019. Caracterización de α - lacta-albúmina : Electroforesis Acrilamida. Objetivos. Distinguir los diferentes geles de poliacrilamida utilizados para estudiar proteínas.

E N D
JA Carde, PhD Modificado de Dra. Liza Jiménez UPR-Ag Lab Biol 4019 Caracterización de α - lacta-albúmina:ElectroforesisAcrilamida
Objetivos • Distinguir los diferentes geles de poliacrilamida utilizados para estudiar proteínas. • Describir la importancia biológica que posee la electroforesis de SDS-PAGE en el estudio de las proteínas. • Caracterizar la proteína α-Lactalbumina a través de electroforesis de SDS-PAGE.
Introducción • Extracción, purificación y caracterización • Para verificar si la proteína obtenida es la deseada. • Pruebas para las propiedades físicas, químicas o biológicas de la proteína. • SDS-PAGE: Forma de caracterizar la proteína
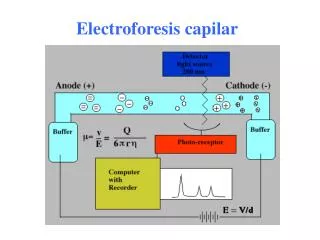
Arne Tiselius 1937 (Nobel 1948) ... en función de su diferente carga eléctrica. Moléc. + cátodo (-) Moléc. - ánodo (+) cátodo SOPORTE ánodo • ELECTROFORESIS DE ZONA • MATRIZ (que retiene las moléculas) + - Introducción Separar y analizar moléculas: Proteínas ADN/ARN ELECTROFORESIS Arne Tiselius 1937 (Nobel 1948)
Electroforésis de Poliacrilamida • Las proteínas y los ácidos nucleicos: moléculas con cargas netas que pueden ser separadas en una matriz gelatinosa utilizando un campo eléctrico (electroforesis). • La velocidad con que se mueven las moléculas sobre la matriz gelatinosa depende de • 1) los parámetros físicos de la molécula (forma, carga) 2) la fricción de la molécula y la viscosidad del medio 3) la fuerza del campo eléctrico
Factoresqueafectan la mobilidad 1 Campo eléctrico Diferencia de potencial (V-voltios): bajo voltaje (10-500 V) alto voltaje (500-10000 V) Intensidad (A-amperios): flujo de carga eléctrica. depende de V y de R. Resistencia: determinada por el soporte. Temperatura: efecto joule refrigerantes 2 Muestra Carga o densidad de carga: carga eléctrica/masa de la molécula Tamaño: debido al poro de la matriz. Forma: forma globular vs lineal: avanza más. 3 Amortigudor pH: determina el grado de solvatación y la carga de las moléculas generalmente es ligeramente básico moléc. - Fuerza iónica: generalmente 0.05 o 0.1 M para que no interfiera 4 Soporte Adsorción Porosidad molecular
Electroforésis de Poliacrilamida • Las proteínas son separadas utilizando geles de poliacrilamida. • Los geles son mezcla de acrilamida y bis-acrilamida. • A esta mezcla se le añade catalizadores del proceso de polimerización • persulfato de amonio (perSO4NH4 • TEMED (N,N,N’N’-tetramethylethylenediamine)
Gel: % vsporosidad • Porosidad∞ %
PAGE – Gel de Poliacrilamida Soporte: (restrictivo) :Poliacrilamida * Polimerización de dos componentes: Acrilamida + Bisacrilamida * Variación de la concentración variación del tamaño de poro Electroforesis vertical Ventajas: * Gran poder de separación(resolución) por CARGA y por TAMAÑO. * Químicamente inertes. * Transparentes (densitograma). * Estables (pH, fuerza iónica, temperatura). * Versatilidad en cuanto al tamaño de poro.
PAGE – Gel de Poliacrilamida • Preparación del gel. • Montaje de la cubeta • Aplicación de la muestra. • Electroforesis. • Detección por tinción: • Azul de coomassie • Sales de plata
Procedimiento: Preparación de la muestra • Coloque 15 µl de la muestra de α-Lactalbumina en un microtubo de 500 µl. • Añada 5 µl de amortiguador de la muestra (Sample Buffer). Este amortiguador contiene mercaptoetanol, un agente que reduce los enlaces de disulfuro en las proteínas, asegurando que se mantiene la configuración helicoidal necesaria para la determinación del peso molecular (figura 4). • Coloque el tubo en un baño de María a 95º C por 5 minutos. • Remueva el tubo del baño de María y colóquelo en hielo mientras prepara el gel de poliacrilamida y el tanque de electroforesis.
Sample Buffer: DTT/B Mercapto DTT/Mercaptoetanol SDS Glicerol Azul Bromofenol Tris-EDTA
Soporte: (no restrictivo) Tiras de acetato de celulosa - + Campo eléctrico (ΔV) • Aplicaciones diagnósticas en serología Electroforesis - Celulosa Soporte: (no restrictivo) Tiras de acetato de celulosa Muestra Electroforesis horizontal Avance de las proteínas • 1. Aplicación de la muestra. • Electroforesis. • Detección por tinción: • Azul de coomassie • Negro amido
Tipos de PAGE’s: SDS vsNativas • Pueden ser desnaturalizados o nativos. • desnaturalizados: se le añade SDS (Sodium Dodecyl Sulfate), detergente con carga negativa. • El SDS al unirse a la proteina la desdobla haciendo la molécula soluble. • El SDS le confiere aTODOS los polipéptidos cargas negativas, la separación de las moléculas es sólo por tamaño • SDS-PAGE (Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis) • Geles nativos no se utiliza SDS la separación…
Electroforesis 2D SDS-PAGE Western Blot Electroenfoque PAGE – Gel de Poliacrilamida • Aplicaciones: • Separar proteínas • Determinar peso molecular de una proteína • Separar proteínas oligoméricas • Separar proteínas en función de su pI • Separar proteínas de mezclas muy complejas • Ver si hay una proteína en concreto
50 kDa S-S S-S S-S - - - - - - - - - - - - - + SDS S-S - - - - - - - - - - - - - 44 kDa S-S - - - - - S-S - S-S S-S S-S - - + SDS + DTT - - - - - - - - - - - - - - - - - 25 kDa PAGE - SDS Condiciones de la PAGE: * No desnaturalizantes: separamos por CARGA y TAMAÑO PAGE *Desnaturalizantes:separación sólo por TAMAÑO SDS-PAGE Agentes desnaturalizantes: SDS Urea Reductores (DTT, b-mercaptoetanol)
Tipos de PAGE’s: Continuidad • Los geles pueden ser continuos o geles discontinuos. (%, pH,) • continuos: la porosidad (PLT el %) es uniforme a largo de todo el gel. • discontinuos: poseen diferentes concentraciones (%) de acrilamida y bis-acrilamida a través de ellos. • “stacking gel”, un gel con poros más grandes; sirve para empaquetar la muestra en un punto específico. • “resolving gel”, un gel de poros más pequeños; produce una mayor resolución en la separación de las proteínas
Proteínas migran hasta la zona del gel donde pH=pI de la proteína + + + pH 2 (ácido) + + pI=6 0 + 0 GRADIENTE DE pH pI=7 0 pH 10 (básico) - - - EnfoqueIsoeléctrico * Punto isoeléctrico (pI) pI es el valor de pH en el que la carga de la proteína = 0 si pH > pI: carga – si pH < pI: carga + * Gel de poliacrilamida (PAGE) con gradiente de pH.
Electroforesis en 2D * 1ra dimensión: ELECTROENFOQUE separa por pI * 2da Dimensión: SDS-PAGE separa por PM
1. SDS-PAGE Electroforesis 3. Incubación con los Anticuerpos 2. Transferencia a membrana proteínas proteínas 2dario proteína interés HRP 1ario Resultado Western BLOT SDS-PAGE 4. Revelado SUSTRATO proteína interés HRP LUZ PRODUCTO Proteína de interés Todas las proteínas Western Blot * Immnoblot interacción PROTEÍNA-PROTEÍNA * Detectar una proteína específica dentro de una mezcla (muy sensible).
Determinación peso molecular • Correr junto a las muestras un marcador molecular estándar (de 8 a 10 proteínas de las cuales se conoce el peso molecular). • Relación lineal entre el logaritmo del peso molecular de las moléculas y la distancia recorrida en el gel. • Crear una curva estándar de la distancia de migración versus el log10 del peso molecular de las proteínas del marcador. • Esta curva se usa para extrapolar el peso molecular de aquellas proteínas en estudio.
Curvaestándar • Para crear la curva estándar es necesario: • 1) Medir la distancia desde el punto de partida (fosas) del marcador hasta cada una de las bandas observadas en el marcador. Se repite lo mismo con las proteínas en estudio. • 2) Determinar el log10 del peso molecular de las proteínas que se encuentran en el marcador estándar y trazar en el eje de Y utilizando papel de gráfica regular. • 3) Trazar la distancia recorrida en el eje de X. Al terminar la gráfica se observa una línea. • 4) Utilizar la distancia recorrida por las muestras y extrapolar a la línea para determinar el peso molecular.
Marcadores de peso molecular: • * Mezcla de diferentes proteínas pre-teñidas de las que conocemos el peso molecular. • * Por comparación y extrapolación podemos averiguar el PM de nuestra proteína. Determinación de Peso Molecular
Preparación del tanque de electroforesis 1. Coloque los platos que contienen el gel de poliacrilamida entre las pinzas que lo sostendrán dentro del tanque de electroforesis. Coloque las pinzas con el gel dentro del tanque. • 2. Añada el amortiguador de la corrida (Running Buffer) al tanque de electroforesis. Asegúrese de cubrir el gel completamente. • 3. Utilizando una pipetta de 1-10 µl, añada 5 µl del marcador estándar a la primera fosa del gel. • 4. Utilizando una pipetta de 10-100 µl, añada los 20 µl de la muestra de α-Lactalbumina (preparada en la parte A) a la segunda fosa del gel. • 5. Continué con el paso 4 para cada grupo de trabajo. • 6. Coloque la tapa del tanque de electroforesis y conéctela a la caja de electricidad. Verifique que los polos están correctamente conectados. • 7. Corra la electroforesis a 100 V por 1 hora.
Tinción de la Gel • 1. Luego de la hora apague la caja de electricidad. • 2. Saque los platos con el gel de poliacrilamida. Con mucho cuidado despegue los platos y remueva el gel. • 3. Coloque el gel en una solución de “Coomassie Blue” y agite por 30-60 minutos a temperatura ambiente. El “Coomassie Blue” es preparado en una solución de ácido acético y etanol, los cuales fijan las proteínas al gel (figura 5). • 4. Descarte el “Coomassie Blue” y lave el gel por 10 minutos con agua destilada. • 5. Utilizando una regla mida las distancias entre las fosas y las diferentes bandas observadas.
Preguntas • 1. Utilizando los datos obtenidos en la parte experimental prepare una curva estándar. Estime el peso molecular de la(s) banda(s) observada(s) en el gel de poliacrilamida. • 2. Al analizar los resultados obtenidos en este ejercicio de laboratorio, ¿Usted puede concluir que los objetivos del pasado ejercicio de laboratorio (modulo 9: Extracción y purificación de proteínas de leche) fueron logrados? Explique su respuesta. • 3. Explique porqué observa más de una banda en la muestra de -Lactalbumina ¿Qué representan las bandas en la muestra de α-Lactalbumina? • 4. Mencione otros métodos que pueden ser utilizados para teñir los geles de poliacrilamida. • 5. Desarrolle una prueba donde se caracterice la α-Lactalbumina en base a sus propiedades físicas, químicas o biológicas.
Quiz 10: Pareo: UnaLetrapor # 1. SDS a. poliacrilamida 2. Mercaptoetanol b. cuál es su tamaño? 3. Comassie c. catalizador polimerización 4. Stacking gel d. azul, buffer de muestra 5. per SO4 NH4 e. romper puentes H |||| O 6. Bromofenol f. cargas negatvias 7. PAGE g. solamente eso 8. Caracterizar h. azul, teñir 9. Purificar i. romper puentes S-S 10.Calor j. gel discontínua