Download

1 / 24
240 likes | 361 Views
C09-15 Impacts de l’imagerie à haut champ (7T et 3T) du tronc cérébral, des noyaux gris et de leurs connexions appliquée aux syndromes parkinsoniens; intérêts pronostiques, physiopathologiques et d’améliorations thérapeutiques. Mise en place «CIC Pitié Salpétrière»

E N D
C09-15Impacts de l’imagerie à haut champ (7T et 3T) du tronc cérébral, des noyaux gris et de leurs connexions appliquée aux syndromes parkinsoniens; intérêts pronostiques, physiopathologiques et d’améliorations thérapeutiques Mise en place «CIC Pitié Salpétrière» Investigateur coordonnateur : Marie Vidailhet Promoteur : Inserm ISP-PRCT Chef de projet : Sébastien Boy ARC promoteur : Pascale Guyot
PROTOCOLE • Objectif principal • Objectifs secondaires • Critère d’évaluation principal • Critères d’inclusion • Critères de non inclusion • Plan expérimental • Prévision des inclusions • Partie réglementaire
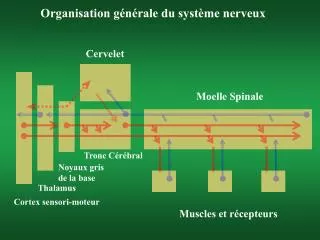
OBJECTIF PRINCIPAL Caractériser de manière précise, par imagerie à haut champ 7 tesla et 3 tesla, la cytoarchitectonique des noyaux des ganglions de la base et du tronc cérébral et de leurs connexions chez le sujet normal et dans les syndromes parkinsoniens
CRITERE D’EVALUATION PRINCIPAL variables mesurées en IRM • La volumétrie des noyaux • La connectivité anatomique des noyaux du tronc entre eux et vers le reste du cerveau • La cytoarchitectonie des noyaux sondée à l’aide de : • L’imagerie de diffusion • La relaxométrie
POPULATION ETUDIEE • Parkinson : pb marche/posture 15 • Parkinson : trouble RBD (sommeil) 15 • Parkinson : aucun des 2 15 • PSP : trouble oculomoteur, marche/posture 10 • Témoins 30 Inscription : VRB : 200-250 euros/patient Période d’inclusion = 2 ans (à partir 1ère inclusion)
CRITERES D’INCLUSIONMaladie de Parkinson (groupe avec RBD) • Homme ou femme de plus de 18 ans • Maladie de Parkinson idiopathique selon les critères de l’UK Parkinson’s Disease Brain Bank • Pour les femmes en âge de procréer : contraception ou test de grossesse négatif • Patient affilié ou bénéficiaire d’un régime de sécurité sociale • Consentement éclairé de l’étude signé • Présence de troubles du sommeil (RBD), en partie détectés par interrogatoire de l’entourage
CRITERES D’INCLUSIONMaladie de Parkinson (groupe avec troubles de la marche et de la stabilité posturale) • Homme ou femme de plus de 18 ans • Maladie de Parkinson idiopathique selon les critères de l’UK Parkinson’s Disease Brain Bank • Pour les femmes en âge de procréer : contraception ou test de grossesse négatif • Patient affilié ou bénéficiaire d’un régime de sécurité sociale • Consentement éclairé de l’étude signé • Présence de troubles de la marche et de la stabilité posturale détectés au best « ON »
CRITERES D’INCLUSIONMaladie de Parkinson (groupe sans RBD ni troubles de la marche et de la stabilité posturale) • Homme ou femme de plus de 18 ans • Maladie de Parkinson idiopathique selon les critères de l’UK Parkinson’s Disease Brain Bank • Pour les femmes en âge de procréer: contraception ou test de grossesse négatif • Patient affilié ou bénéficiaire d’un régime de sécurité sociale • Consentement éclairé de l’étude signé • Absence de troubles du sommeil (RBD) et de la marche et de la stabilité posturale
CRITERES D’INCLUSIONMaladie de Steele-Richarson (PSP)° • Homme ou femme de plus de 18 ans • Maladie de Steele-Richardson (PSP) selon les critères cliniques du National Institute of Neurological Disorders and Stroke (NINDS) modifiés (instabilité posturale et chutes dans les 3 ans suivant le début de la maladie, au lieu de 1 an comme initialement décrit dans les critères initiaux) • Pour les femmes en âge de procréer : contraception ou test de grossesse négatif • Patient affilié ou bénéficiaire d’un régime de sécurité sociale • Consentement éclairé de l’étude signé
CRITERES D’INCLUSION Témoins • Homme ou femme de plus de 18 ans • Examen neurologique normal • Pour les femmes en âge de procréer : contraception ou test de grossesse négatif • Sujet affilié ou bénéficiaire d’un régime de sécurité sociale • Consentement éclairé de l’étude signé
CRITERES DE NON INCLUSIONPatients et témoins • Age <18 ans • Patient protégé, sous tutelle ou curatelle • Contre indications à l’IRM • Pathologie psychiatrique évolutive • Pathologie évolutive engageant le pronostic vital à 1 an • Déficience du patient rendant difficile, voire impossible, sa participation à l’essai ou la compréhension de l’information qui lui est délivrée • Femme enceinte ou allaitante • Femme en âge de procréer sans contraception efficace • Présence d’un trouble de la marche non lié au syndrome parkinsonien • Pour les témoins uniquement : troubles de la marche ou de la stabilité posturale
PLAN EXPERIMENTAL • Patients parkinson • Avec ou sans RBD Pré sélection : Critères Contre indications CE Inclusion : examen clinique, ATC, … évaluation neuro-psy questionnaires enregistrement sommeil J2 : IRM 3T CENIR OFF puis ON enr oculaires J3 : tests marche,… IRM 7T : Neurospin J2 J2 J3 J-90 J1 + appel téléphonique 1 semaine après J2
PLAN EXPERIMENTAL Patients PSP Témoins • + appel téléphonique 1 semaine après J2 Pré sélection : Critères Contre indications CE Inclusion : examen clinique, ATC, … évaluation neuro-psy questionnaires IRM 3T CENIR enregistrement sommeil • J2 : • tests marche,… • enr oculaires • IRM 7T : Neurospin J1 J-90 J2
PARTIE REGLEMENTAIRE • Participant :Information et consentement • Données : Anonymisation, Support de recueil de données, Recueil et traitement • Vigilance : Définition et déclaration des EIG • Suivi / Qualité : Centre investigateur, pharmacie, laboratoire • Modifications substantielles
INSCRIPTION CLINICAL TRIAL • Inscription sur le site clinicaltrials.gov • Inscription avant la 1ère inclusion → pour publications En cas de difficultés, contacter Béatrice Barraud (responsable qualité en recherche clinique) Tél : 01 44 23 67 29
INFORMATION DU PARTICIPANT • Information (NI : 1ère partie du formulaire) • Orale + écrite • Donnée par investigateur • inscrit à l’ordre des Médecins • déclaré au promoteur • Notice d’information paraphée par le participant en bas de chaque page • Avant tout acte pratiqué dans le cadre du protocole de la recherche
CONSENTEMENT Consentement (CS : 2ème partie du formulaire) • Formulaire daté et signé par : • La personne qui se prête à la recherche • L’investigateur qui recueille le consentement • 3 exemplaires originaux : • 1 ex. remis à la personne • 1 ex. conservé par l’investigateur • 1 ex. pour le promoteur • Date de signature = début de participation à l’étude • Date de signature ≤ date du premier acte pratiqué Inscrire dans le dossier source (dates d’information et de signature) « Dés la 1ère inclusion, envoyer l’information au promoteur puis point mensuel inclusions »
ANONYMISATION DES DONNEES • Supprimer tout élément nominatif des copies documents sources à adresser au promoteur excepté sur le consentement éclairé : • Support de recueil des données cliniques et biologiques • Copies d’examen, compte – rendus … • Attribuer un code anonyme à chaque participant : Ex : 1ère lettre du nom + 1ère lettre du prénom + l’année + N° inclusion • Inscrire la correspondance code anonyme / identité : Liste des participants (document promoteur unique confidentiel)
SUPPORT DE RECUEIL DE DONNEES Pour assurer la qualité de gestion des données : • Remplissage du CRF • Écrire au stylo à bille noir en lettres capitales • Remplir toutes les cases : • DM=donnée manquante • NF=non fait • NA=non applicable • NC=non connu • Reporter le code du participant sur chaque page • Correction du CRF • Rayer d’un simple trait • Corriger la donnée • Dater et parapher par l’investigateur Attention : le CRF n’est pas un dossier source
DEFINITION DES EIG • Tout événement ou effet indésirable qui : • Entraîne la mort • Met en danger la vie de la personne qui se prête à la recherche • Nécessite une hospitalisation ou une prolongation de l’hospitalisation • Provoque une incapacité ou handicap important ou durable • Se traduit par une anomalie ou une malformation congénitale • Autres cas : • Événement nécessitant intervention médicale ou chirurgicale pour prévenir l’évolution vers un des états précités, et certains résultats paracliniques • Tout événement ou résultats potentiellement grave selon le jugement de l’investigateur ou du promoteur
DECLARATION DES EIG Pour tout EIG, l’investigateur doit : • Noter l’événement dans le CRF • Évaluer la gravité et le lien de causalité (EIG/DM ou les procédures de mise en œuvre du DM ou le(s) traitement(s) associé(s)) • Compléter le formulaire de déclaration initiale d’EIG Au minimum : nom et coordonnées de l’investigateur, numéro de l’ essai, numéro de patient, nature de l’ EIG (diagnostic), Produit, critère de gravité, lien de causalité Documenter l’événement à l’aide de comptes-rendus d’examens, d’hospitalisation… • Envoyer, dans un délai de 24h maximum à partir de la constatation de l’EIG, le formulaire de déclaration et les éléments de documentation anonymisés au promoteur par fax Suivre l’événement jusqu’à complète résolution ou stabilisation Envoyer des formulaires de déclaration complémentaire
SUIVI / QUALITE • Monitoring en cours d’étude en présence de l’investigateur : • Après la première inclusion • Régulièrement selon la vitesse d’inclusion • En fin d’étude • Audit / Inspection : • A tout moment au cours de l’étude • Jusqu’à 15 ans après la clôture de l’étude • Archivage des documents pendant 15 ans après la clôture de l’étude
SUIVI / QUALITE • Monitoring / Audit / Inspection de l’étude d’après les documents suivants : • Consentement de tous les participants • Supports de recueil de données (CRF) • Dossiers source de tous les patients ayant signé un consentement • Classeur investigateur tenu à jour • Tout document concernant les produits à l’essai et les échantillons • En présence obligatoirement de l’investigateur pour : • Fournir les informations nécessaires • Répondre aux questions médicales éventuelles • Réaliser les corrections nécessaires datées et paraphées
MODIFICATIONS SUBSTANCIELLES • Modifications substantielles du protocole en cas de : • Ouverture de centre • Changement d’investigateur • Ajout d’examens • Changement d’objectifs • Prolongation de la durée de l’étude • Augmentation du nombre d’inclus • Modifications de critères d’inclusion, de population • …… • Avertir le promoteur • Attendre l’accord du promoteur avant de tenir compte des modifications • Attention 1 modification substantielle / 6 mois sauf cas exceptionnels • Comme convenu, une modifications substantielle est prévu dès la 3eme inclusion afin de mettre à jour le protocole concernant les examens.