Download

1 / 41
450 likes | 804 Views
Vibrational Spectroscopy. Observe transitions between vibrational levels of molecules Changes in rotation as well; Ro-vibrational spectra Observed as: one photon absorption & emission (mostly in mid-IR spectral region) As inelastic scattering of light (Raman)

E N D
Observe transitions between vibrational levels of molecules • Changes in rotation as well; Ro-vibrational spectra • Observed as: • one photon absorption & emission (mostly in mid-IR spectral region) • As inelastic scattering of light (Raman) • As nonlinear 4-wave mixing (CARS) • Universally applicable. • All polyatomic and heteronucleardiatomics have at least one IR allowed fundamental transition • All molecules have a Raman vibrational spectra.
Vibrational spectroscopy can be applied to liquids, solids, and surfaces as well as in gas phase. • There is a high degree of qualitative chemical information in a vibrational spectrum • Different types of chemical bonds lead to IR absorption bands in predicable spectral intervals. • Bernath has a table of such IR frequencies • These are really wavenumbers not frequencies • In “finger print region” the normal modes are quite mixed but can complex spectra can be used for identification
Modern electron structure calculations can predict vibrational spectra with remarkable accuracy for most molecules • B3LYP DFT is widely used; after empirical scaling errors of a few % are typical. • IR spectroscopy of rare gas matrices have given the first spectroscopic evidence for many unstable and highly reactive species • IR spectra provides window into Intramolecular dynamics – how energy deposited in one mode can flow to other modes. • These “IVR” processes are fundamental to most chemical reactions.
Once vibrational spectra are assigned, they can be used to selectively detect molecules • IR absorption typically stronger an MW absorption • For OCS strongest IR integrated line strength ~2000x larger than for strongest rotational transition at room T. • FTIR’s are widely used for chemical process control • IR spectroscopy can be used to probe with ~5mm micron spatial resolution • Raman & CARS microscopy can be extended down to ~200 nm resolution • IR spectroscopy can follow population changes into the fsec time domain • Time vs. Freq. resolution trade-off.
Theory Foundations • Vibrational spectroscopy based upon Born-Oppenheimer approximation • Vibrational states found by solving SE for nuclear motion on potential surface generated by electronic motion. • Almost all IR absorption and emission bands arise from electric dipole transition moments • Raman transitions arise from vibrational dependence of molecular polarizability
How do we solve the vibrational SE? • For diatomic molecules, vibrational energies and wavefunctions can be solved essentially exactly at trivial computational cost by numerical integration • For small polyatomic molecules (3-5 atoms), vibrational states can be found by variational calculations in a large basis set. • For most larger molecules, vibrational motion treated by (quasi degenerate) perturbation theory calculations • Double perturbation theory: Anharmonicity & vibration-rotation interactions.
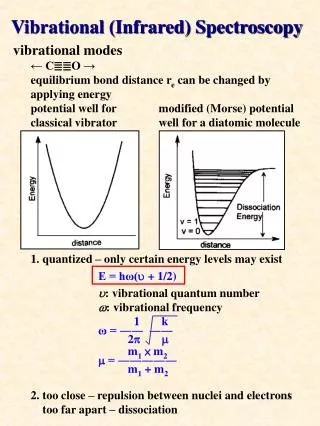
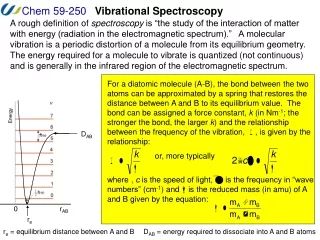
Diatomic Molecules • Wavefunctions are produce of spherical harmonic function (rotational wavefunction) times a radial function c(r). Radial SE: • With c(0)=0. Probability density |c(r)|2
Harmonic Oscillator – Rigid Rotor • Expand • Keep only harmonic (k2) term. • Replace • Ignore fact that r does not go to minus infinity • We can now solve this problem by elementary quantum mechanics
Vibrational Spectroscopy II:Polyatomic molecules Key reference sources: E.B. Wilson, J.C. Decius, and P.C. Cross, Molecular Vibrations: Theory of Infrared and Raman Vibrational Spectra (Dover) G. Herzberg, Molecular Spectra and Molecular Structure: II Infrared and Raman Spectra of Polyatomic Molecules
Starting point is Harmonic theory of vibrations • Even when we want higher accuracy, we almost always start from the Harmonic theory and do either perturbation theory or variational calcs. • We will start with analysis of purely vibrational motion and then go on to ro-vibrational spectroscopy
Harmonic expansion • Let us have N atom molecule with equilibrium positions of atoms xie, yie , zie (i = 0….N-1) and instantaneous positions xi, yi , zi. • Define mass weighted coordinates by: • In terms of these coordinates the Kinetic Energy is given by:
Potential Energy There is no term linear in q’s because we expanded around the equilibrium position. In Harmonic theory, we truncate after quadratic terms. f is a symmetric matrix with positive definite eigenvalues (or else the re position would be a saddle point instead of an equilibrium positions. Thus we can write the eigenvalues as wi2 with orthonormal eigenvectors lji. Define normal modes of vibration Qi by:
Classical Solutions • Qi (t) = Ki cos( ωi t + fi) so wi is the angular velocity of i’th normal mode. Divide by 2πc to get wavenumber. • Define dipole derivatives • To first order in q (harmonic approximation for dipole moment)
Classically, an oscillating dipole moment radiates at its frequency of oscillation. Thus, we can expect a normal mode will produce emission (and thus by the Einstein relations, absorption also) when at least one component of mi is nonzero. • Polarizability derivative • Normal mode will produce Raman scattering with wavenumber shift wi/2πc if at least one component of aiis nonzero
Zero frequency modes • For a nonlinear (linear) molecule, there will always be 6 (5) normal mode vectors l that have zero eigenvalues. 3 of these will correspond to the x,y, and z translation of the entire molecule, which can have zero restoring force. 3(2) correspond to infinitesimal rotation of the molecule, which also has zero restoring force.
Symmetry Analysis • Each normal mode is part of irreducible representation of the Point Group of the molecule’s equilibrium structure. • Form a 3N dim. Rep. from the x,y,z displacement coordinates of each the N atoms • Reduce this into the irreps. • Subtract irreps for x,y, and z (for center of mass motion) and Rx, Ry, and Rz (for rotations) • The remaining irreps give the distribution of the normal modes among symmetry classes. • The number of totally symmetric modes equals the number of unique structural parameters of equilibrium structure.
Symmetry (cont.) • Normal modes which have the symmetry of x,y, or z are IR symmetry allowed. • The fundamental transition will have components of the dipole matrix element that have same symmetry as the Cartesian components. • Normal modes which have symmetry of x2, y2, or z2 have polarized Raman scattering fundamentals • Those with symmetry of xy, yz, or xz have depolarized Raman scattering fundamentals.
Quantum Solutions • Let Pi be momentum conjugate to normal mode Qi.
IR & Raman Selection Rules • If we approximate: • (double harmonic approximation) • Qi have nonzero transition matrix elements between states with Δni = ±1 while all other Δnj =0. (Fundamentals) • Same Δni = ±1 selection rule applies to Raman Spectroscopy if we truncate polarization expansion at linear terms. • Exclusion Rule: For molecules with center of inversion, IR allowed fundamentals are all of u symmetry, while all Raman fundaments are g.
Matrix Elements In general Qm has matrix elements with Δn = -m, -m+2….+m. Similarly for Pm. Q can be written as sum of raising and lowering operators. P as i times the difference.
Overtones and Combination bands • Spectra show transitions from ground state to states with multiple quanta in one mode (overtones) or multiple modes (combination bands) principally for two reasons • The dipole (and polarizability) are not simply linear functions of the normal modes, but have higher order terms in their expansions. The corresponding orders in the expansions allow transitions with the same order changes in vibrational quantum number
Overtones and Combinations (cont). • The potential has higher order terms beyond harmonic terms. • For stretching vibrations, the potential is more like Morse Oscillator which has rapidly decreasing but nonzero matrix elements for all Δn. • Off-diagonal terms such as Qa2Qb, will mix the state with nb = 1 with the one with na = 2. This allows the state with na = 2 to “borrow” or “steal” intensity from the nb = 1 fundamental. • This is known as Fermi resonance when third order term is involved and is quite common.
Internal coordinates • Potential energy surface much easier to parameterize in terms of bond lengths and bond angles rather than atomic Cartesian coordinates. • We can use internal coordinates for vibrational problem. • In general, these internal coordinates are curvilinear and give very complex kinetic energy operators. • More common to define rectilinear internal coordinates that have atoms move in straight lines. • Minimum energy to bend is an arc that is somewhere between rectilinear motion and constant bond length bend. • These internal coordinates are not “orthogonal” which introduces come complications.
Internal coordinates (cont) • Commonly used internal coordinates are: • bond lengths: rij = |ri – rj| • bond angles qijk equal to the angle between unit vectors j->I and j->k • and torsional angles tijkl equal to the angle between planes formed by atoms i,j,k and that formed by atoms j,k,l • Out of plan distance h, equal to the distance of an atom i from the plan formed by atoms j,k,l
For each internal coordinate, St, define a 3N vector Bt which has components equal to the derivatives of St with respect to the Cartesian coordinates of the atoms. The set of column vectors Bt make a matrix B. • Rectilinear coordinates defined by St = Si BtiΔxi • Explicit expressions for B elements given in Wilson, Decius, and Cross
Define G matrix by • Kinetic energy: • Define F matrix elements by
The square of the vibrational angular frequencies are the eigenvalues of the matrix G.F. • Note that even though G and F are symmetric matrices, their product is not (since they do not commute) • Eigenvalues are still real and positive definite or stable minimum energy point. • Define matrix, L, of right handed eigenvectors by • G.F.λ = L.λ, where λ is diagonal matrix of eigenvalues • L normalized by Lt G-1 L = I (identity matrix), Lt FL = λ • This matrix is not orthogonal L-1 = Lt G-1. • S = L.Q, Q = L-1.S, Ps = (L-1) t.PQ PQ = L t.PS • L matrix is real which means all atoms reach end points of vibration at same time (though for asymmetric modes, some could be at outer turning points when others at inner turning pts.
Redundant Coordinates • It is often useful to define more internal coordinates than there are normal modes because of angle constraints. • For planer molecule with 3 atoms bound to common atom (for example H2CO, the sum of the three bond angles = 2p. • For 4 bonds from common atom (often C), there are six bond angles, but only 5 are independent. • For ring compounds, the sum of the internal bond angles are p (N-2). • We could use constraint to express one angle in terms of the others, but that will kill the symmetry. • If we ignore the constraints, but put in physically realizable force constants, we will get zero eigenvalue for normal mode that is perpendicular to constraint condition.
Symmetry Coordinates • We can do a linear transformation of the internal coordinates to produce a set of symmetry coordinates that transform as irreducible reps of the point group. • We can use symmetry projection operators to generate these from the internal coordinates. • Each symmetry coordinate will be linear combinations of symmetrically equivalent internal coordinates (i.e. those that transform into one another) • If we transform the F and G matrices by the matrix that transforms to symmetry coordinates, they will each block diagonalize into symmetry blocks. Same for FG product.
Isotopic Substitution • The shift in vibrational spectrum with isotopic substitution is often used to assign character of normal modes. • By first order perturbation theory: • Thus the shift gives us the kinetic energy of the substituted atom in each normal mode. • Redlich-Teller Product rule: (WDC sect. 8.5)
Vibrational Angular Momentum • Define Vibrational Angular Momentum operators (Pa):
Wilson –Howard Hamiltonian (1936) • This is so complicated that it is almost impossible to use without further simplification!
Watson HamiltonianMol. Phys. 15, 479-90 (1968) • James Watson showed he could dramatically simplify the Wilson-Howard Hamiltonian The U term (which arises from K.E.) depends only on Q’s and thus is a like a mass dependent kinetic energy term. The Watson Hamiltonian has been used to do essentially exact variational calculations of ro-vibronic energy levels of polyatomic molecules.
Parallel transitions (DK = 0) The P,R branches follow polynomial in m expression as for linear molecule but with |m| > K. Honl-London Factors
Honl-London Factors PP and RR branches (ΔK = ΔJ) are strong, RP and PR (ΔK = -ΔJ) weak