Download
1 / 39
400 likes | 655 Views
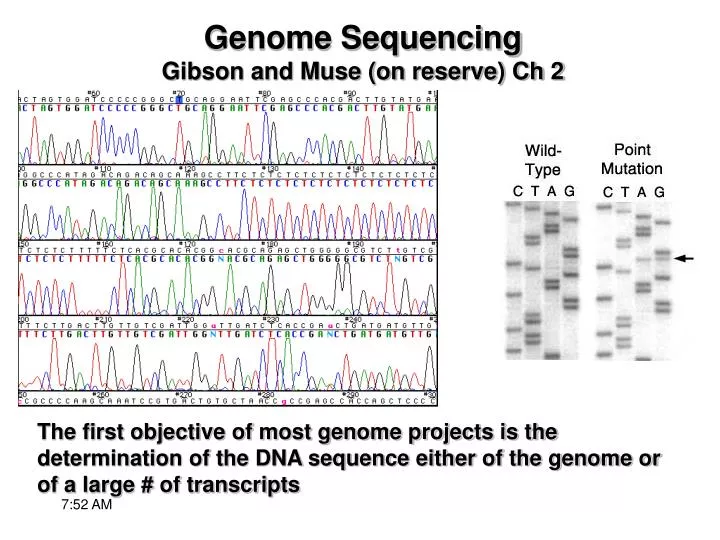
Genome Sequencing Gibson and Muse (on reserve) Ch 2. The first objective of most genome projects is the determination of the DNA sequence either of the genome or of a large # of transcripts. Most current genome projects rely on chain termination sequencing developed in 1974 by Frederick Sanger

E N D
Genome SequencingGibson and Muse (on reserve) Ch 2 The first objective of most genome projects is the determination of the DNA sequence either of the genome or of a large # of transcripts
Most current genome projects rely on chain termination sequencing developed in 1974 by Frederick Sanger The basic idea behind Sanger Sequencing is to generate all possible single-stranded DNA molecules complementary to a template The template starts at a common 5’ base and extends up to 1.5 kb Sanger (di-Deoxy) DNA sequencing
ssDNA fragments are labeled to allow identification of the 3’-most base Fragments are separated by size by gel electrophoresis Resulting ladder of fragments is then read Sanger (di-Deoxy) DNA sequencing
Template can be cloned DNA, genomic DNA or PCR product DNA polymerase, primer (3’OH) and dNTPs are used to synthesize DNA Sanger (di-Deoxy) DNA sequencing
Dideoxymethod uses DNA bases containing modified deoxyribose sugars = dideoxyribose which contain H at the 3’ position of the ribose sugar rather than OH Modified sugars cause chain termination Sanger (di-Deoxy) DNA sequencing
ddNTPs can be labeled w/ 35S and run as 4 different rxs (~500bp) ddNTPs can be labeled w/ fluorescent dyes and run as 1 rx (~1kb) Fragments are separated by differential retardation of migration of molecules of different size Sanger (di-Deoxy) DNA sequencing
Improvements on Sanger Sequencing • Four-color fluorescent dyes, dye-terminators, also dye-labeled primers (req.s 4 lanes) • Seq. rx products are “laser” scanned just before running off an electrophoresis medium - reading is automatic • Improvements in chemistry and bioengineered DNA polymerase - better handling of secondary structure, longer (~1.2kb) reads • Replacement of slab gels w/ capillary electrophoresis - longer reads, no gel pouring - the art of pouring gels is dead • In total, ~20x increase in sequencing productivity
New Sequencing Methods - SBH Emerging technologies will reduce the cost and time associated with genome sequencing Sequencing by hybridization (SBH) - uses complementarity between 2 strands to detect whether an exact match to an oligo is present in a sample of DNA. This can be used in “re-sequencing” looking for SNPs, e.g. a variant detector array (VDA) contains all possible oligionucleotides in a molecule several kb in length as well as mismatches at each sites - look for binding of sample to different spots in the array This technique is being used to resequence primate genomes and scan for 1.5 million SNPs in human genome
New Sequencing Methods - Mass Spec. Mass Spec. techniques have been used to seq. fragments up to 50 bp, identity assigned by time of flight through a vacuum chamber. In principle, it should be possible to determine seq. of molecule that is devided into all possible oligonucleotides
New Sequencing Methods - nanopore technology Nanopore sequencing strategies - one approach is to use monitor electrical current as a single strand of DNA or RNA passes through a pore in a membrane, another approach channels nucleic acid through a single-molecule fluorescence reader These methods have the potential of reading 100s of kb of sequence in minutes
New Sequencing Methods - single-molecule Sanger Some single-molecule approaches using the Sanger method focus on detecting fluorophore incorporation into single molecules on an array 454 Corporation machines amplify single molecules using massively parallel picoliter scale amplification and detection “Pyrosequencing” platform GS20 - DNA sequence is determined by analyzing flashes of light released by release of pyrophosphate with chain extension using a predetermined sequence of nucleotide addition in picoliter-sized reactions
Typing HPV by Pyrosequencing Pyrosequencing, a bioluminometric DNA sequencing technique, is replacing Sanger Sequencing in some applications
Typing HPV by Pyrosequencing Pyrogram showing genetic variation between strains
New Sequencing Methods - pyrosequencing A single run GS 20 generates up to 25 million high-quality bases in hundreds of thousands of short sequences(~100bp) 2 sequencing runs produced 99.75% of 2 chloroplast genomes (~150kb each) to 25x and 17x coverage w/ error rates tested against conventional sequencing of ~0.04%
New Sequencing Methods - pyrosequencing Comparison of Sanger, 454, Illumina, and ABI SOLiD
Reading Sequence Traces • Base-calling is routinely done by automated software that reads bases, aligns similar sequences and allows easy editing of sequences • phred - software that converts traces into sequence data that can be deposited into database, including probability scores for each base call • 4 fluorescent spectra are merged, maintaining peak register • Mean peak distance is used to determine where peaks should be - insert “n”s for missing peaks, call doublets as single bases (heterozygotes) • Adjust peak distance for changes as run proceeds and for variation in GC content • <1 second required to call ~1kb
Reading Sequence Traces 1st ~50 bases “messy” SNPs and indel between two sequences Traces also become difficult to read at end of seq. - diffusion and sm. relative difference in size of fragments Depending on end use, seq.s can be manually edited
Reading Sequence Traces - Errors All automated base-calling algorithms make errors, so we assign probabilities to each call phred uses 4 parameters The variance in peak spacing over a 7-peak window centered on called base The ratio of the largest uncalled to the smallest called peak in that window The same ratio over a 3-peak window The # of bases between the current base and the nearest unresolved one
Reading Sequence Traces - Errors The error probability, p, is converted to a phred score, q = negative logarithm of p, q < 13 means that greater than 0.05 probability that the base is miscalled q>30 means an error probability of 0.001 q > 20 is regarded as high-confidence Bases much further from the start get progressively smaller phred scores
Contig Assembly • Sequences of DNA fragments longer than ~1kb have to be assembled - aligned and corrected - again often done w/ software, e.g. phrap assembler and consed graphic editor, general features: • Color highlighting of features such as different bases, quality scores, regions of seq. conservation, manual vs automated calls • Ability to view and work between actual traces and edited seq. • Display of complementary strand • Manual editing tools - insertion/deletion etc, linked to output • Alignment algorithm • Computation of probability scores for consensus • Ability to id potentially polymorphic sites
Contig Assembly Editing starts w/ chromatogram files which are generally left untouched Base call files (phred) include quality scores, base calls, peak positions Assembly files (phrap) include alignment, consensus contig seq. and score Additional flies can be saved after editing, etc Averaging across multiple reads provides the high confidence in automated DNA sequence determination
Genome Sequencing 2 traditional approaches to genome sequencing Hierarchical - ordered, start w/ low res. physical alignment Shotgun - break the genome into small manageable pieces
Hierarchical Sequencing Earlier form, aka - Top-down, map-based, clone-by-clone Efficient use of sequencing and computational resources, fosters high-res. physical and genetic maps and allows sharing of work across consortia First step is cloning fragments into manageable units
DNA Libraries Digested or sheared genomic or chromosomal DNA is ligated into the MCS of a vector Aim for 5-10x redundancy - every region represented 5-10 times Select clones by matching end seq. to reconstruct entire chromosome = tiling path
DNA Libraries - hybridization Chromosome walking uses restriction fragment lengths - restriction profiling - and hybridization using labeled probes to order clones
Clones can be ordered by comparing restriction digest profiles using alignment software DNA Libraries - Fingerprinting Restriction digest of clones = finger print All cones that share the labeled probe seq. Band profile – shared bands, vector band, unique bands 1,3,4 form a tiling path
DNA Libraries - End-sequencing Gaps remaining after fingerprinting can be filled by sequencing both ends of a collection of BAC clones and comparing w/ already the sequenced genome - finding a match to one end implies that the clone covers a gap Once a tiling-path is chosen, BAC clones are sheared into small fragments that are subcloned for automated sequencing, often into phagemid vectors (hold 1kb of DNA) or plasmid (2-3kb) and seq. both ends Enough seq. reactions are conducted to ensure ~10x coverage Remaining (small) gaps are filled in the sequence “finishing” phase
Shotgun Sequencing Computer algorithms are used to assemble contigs derived from 100,000s of overlapping seq.s Construct plasmid library from whole genome and seq. to achieve 5-10 fold redundancy Multiple sequences are aligned using algorithms that screen out repetitive sequences This assembly can resolve ~90% of the genome - filling in gaps can take as long as initial phase
Shotgun Sequencing - software Screener - marks and hides (masks) seq. containing repetitive DNA (e.g. microsatelites, LINEs, ribosomal DNA) Seq. are not removed, but screened from alignment algorithm so they do not contribute to overlap - length and location is taken into account in genome assembly Overlapper - compares all the reads searching for overlaps of predetermined length and identity, e.g. 40bp overlaps w/ no more than 6 errors - 1 in 1017 by chance “errors” account for seq. errors, polymorphisms, heterozygotes Parallel processing on 40 supercomputers, each w/ 4Gb of RAM overlapped 27 million human seq. reads in <5 days
Shotgun Sequencing - unitig Unitigger - resolves repeat-induced overlaps due to low copy number repeats (tandem or dispersed duplicates) A unitig is a contig formed w/ a series of overlapping, unambiguously unique sequences
Shotgun Sequencing - overcollapsed unitig Overcollapsed unitigs are repetitive elements that have been incorrectly collapsed into a single unitig Overcollapsed unitigs can be identified b/c they appear to have a much higher level of coverage (b/c of repeats through genome) than the rest of the seq.
Shotgun Sequencing - coverage Highly likely unitigs (U-unitigs) formed from 6x coverage can cover up to 98% of nonrepetitive euchromatin in stretches of unique DNA greater than 2kb in length = 75% of the human genome
Shotgun Sequencing - software Scaffolder - uses mate-pair information to link U-unitigs into scaffold contigs HGP and D. melanogaster project primarily seq.ed from both ends of 2kb or 10 kb clones - these were correctly paired ~98% of the time Contiguity of assembled scaffold was verified using 50kb mate-pairs
Shotgun Sequencing - gaps Many remaining gaps are from repeats - and many can be resolved by computationally aggressive, but more error-prone, techniques The level of coverage must balance genome size, funds and needs of project
Shotgun Sequencing - gaps 3 approaches to filling gaps If gap from removed repeat, seq. can be reinserted as a generic sequence - no polymorphisms etc It may be possible to find a cloned seq. that bridges gap and seq. that (e.g. 50 kb clone instead of 2 or 10kb clones) Gaps may be bridged using seq. from different projects e.g. cDNA seq., comparison w. other species in search of loci that may bridge gap (design PCR primers to direct seq.ing) Finally scaffolds are assembled into chromosomal locations, compare with genetic map or place on cytological maps w/ fluorescent in situ hybridization (FISH)
Sequence Verification Whole genome seq. must be assessed at 3 levels: 1. Completeness. Microbial genomes seq.ed for single isolates in entirety, may contain ~kb gaps Most higher eukaryotic genomes contain heterochromatin that may never be seq.ed, except in small islands 2. Accuracy. Assessed by probability scores, can be increased w/ greater coverage (overall or in spots) 3. Validity of assembly. Assessed by internal consistency or contrasting w/ pre-existing genetic or physical maps. Can compare w/ predicted restriction profiles w/ observed fingerprints, and spacing of paired end seq.s
At time of publication IHGSC and Celera human genomes, draft genomes, contained hundreds of inconsistencies and gaps Only chromsomes 21 and 22 were “finished” and these contained far fewer problems = differences likely due to assembly Sequence Verification