Download
1 / 8
80 likes | 205 Views
IB404 - 15 - Human genome 1 – Mar 7. 1. Many studies on human chromosome structure have been done, and of course the karyotype is well known, from a large variety of visualization methods. We have 23 different chromosomes, with each diploid cell having 46 total. X and Y are homologs.

E N D
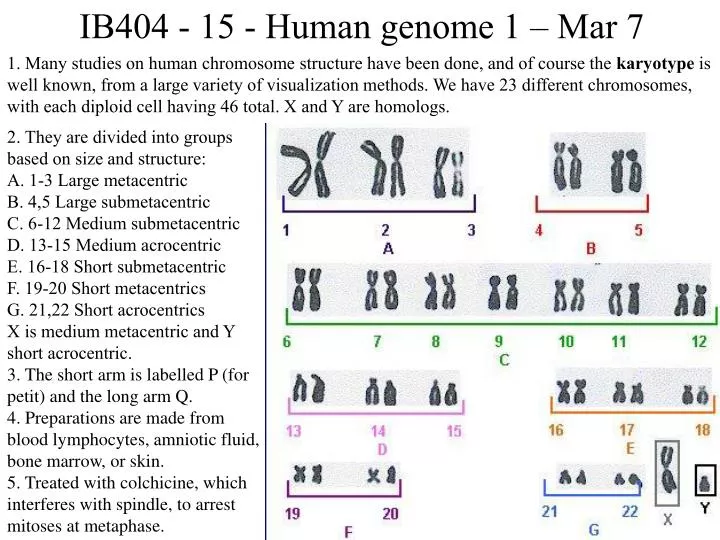
IB404 - 15 - Human genome 1 – Mar 7 1. Many studies on human chromosome structure have been done, and of course the karyotype is well known, from a large variety of visualization methods. We have 23 different chromosomes, with each diploid cell having 46 total. X and Y are homologs. 2. They are divided into groups based on size and structure: A. 1-3 Large metacentric B. 4,5 Large submetacentric C. 6-12 Medium submetacentric D. 13-15 Medium acrocentric E. 16-18 Short submetacentric F. 19-20 Short metacentrics G. 21,22 Short acrocentrics X is medium metacentric and Y short acrocentric. 3. The short arm is labelled P (for petit) and the long arm Q. 4. Preparations are made from blood lymphocytes, amniotic fluid, bone marrow, or skin. 5. Treated with colchicine, which interferes with spindle, to arrest mitoses at metaphase.
6. Separating the similar size and centromere chromosomes requires staining, for example, with Giemsa, yielding G-banding unique to chromosomes. Giemsa differentially stains AT-rich regions, with about 300 major bands in the human genome. On the left is what a cell squash at metaphase looks like, on right is one chromosome, with G-banding, p/q units, and marker genes.
7. There had, of course, been an enormous effort towards cloning human genes over the past 30 years. As was true for most animals, these efforts were primarily directed at cloning and sequencing cDNAs, thereby avoiding the typically long introns of human genes. Some 10,000 different genes had been characterized in this way by 2000. 8. Back around 1990 a serious effort was initiated to plan and start the human genome project, under which umbrella the C. elegans and D. melanogaster projects were supported as pilots. The Department of Energy actually got started first, however NIH soon took over with James Watson of Cold Spring Harbor as the first leader of this effort. The basic plan was 15 years and $5b, with faith that the technology would improve and costs would come down. It cost $3b and 12 years. 9. The public project was based on a physical map of YACs and BACs, because the known complexities of thousands of copies of transposons was thought to make any other approach untenable. The physical map was completed in 1995 and serious sequencing of clones started around then at many centers, including the Big Three in the US, that is MIT Whitehead in Boston under Eric Lander (left), WashU in St. Louis under Bob Waterston (next), and Baylor College of Medicine in Houston under Richard Gibbs (next). The Sanger Center in Cambridge under John Sulston (next), and additional efforts at RIKKEN in Japan and the BGI in China contributed.
10. In 1998 Craig Venter (left) founded Celera Genomics with $300M from Applied Biosystems Inc (ABI) including 300 of their new 99-capillary Sanger sequencers. The plan was a WGS of the human genome after demonstrating that it could work on D. melanogaster. The intention was to complete it by 2001, four years ahead of the public project. This galvanized the public project, which massively increased their technologies to similar levels including major automation with robotics, and the concentration of effort in the Big Three NIH-funded sequencing centers in the US, the Joint Genome Institute (JGI) at DOE, and the Sanger Center at Cambridge in England. Under the guidance of Francis Collins (right) heading the National Human Genome Research Institute at the NIH (now the NIH director), they decided to draft-quality sequence their BACs instead of finishing them, to compete with Celera. The competition between these two groups became particularly intense and ugly, until eventually the director of genome sequencing at DOE, Ari Patrinos (middle), was asked by president Bill Clinton to negotiate an agreement to co-announce their draft sequences in June 2000 at the White House with Tony Blair, and they published papers in February 2001, in Science for Celera and Nature for the public project. Celera shares 2000 2004
11. The détente was only superficial, however, and these two groups have continued to fight with each other ever since. This schism has even led to most public projects being routinely published in Nature, with Celera and other industry papers appearing routinely in Science, until recently. 12. The public consortium insisted that Celera cheated by using the public sequence data in a way that retained a lot of the assembly information, while Venter insisted that if Celera had finished the job they would have had a far better product. As it is, Celera fired Venter and abandoned sequencing, while the public project finished the sequence in 2003, in time for the 50th anniversary of the publication of the Watson and Crick double-helix structure for DNA in Nature. 13. In the end of the day, as is so often the case, both groups were right. WGS is clearly the most efficient way to sequence genomes, even such large and complex ones, and the public projects have now used WGS approaches in various ways for the mouse, rat, and other mammalian genome projects, let alone fungi, insects, and worms. But the model of performing such projects in the public domain and making all information freely and rapidly available is clearly the way to go, as argued most forcefully by John Sulston and others, including Fred Sanger. 14. Even Venter has come around and now has DOE support to sequence environmental samples for bacteria and protists, and even NIH funding for individual genome projects. NIAID separately funded the Anophelesgambiae genome at Celera, and TIGR (which later became the J. Craig Venter Institute or JCVI) has sequenced many others like the Asian tiger mosquito Aedesaegypti (vector of dengue fever virus - ±1Gbp) and the house mosquito Culexpipiens (vector of West Nile virus - ±500Mbp). Today Venter’s main projects have moved on to synthetic biology, trying to construct novel bacteria to do specific things, like generate novel fuels.
15. Whose genome was sequenced? The public project used genomic DNA from 10 different anonymous donors to build their YAC and BAC libraries. Celera claimed to have used 5, but in fact about 75% of the sequence is Venter’s, and he subsequently paid about $100m to finish his. Humans are such a young species that we differ from each other, and indeed the two genomes in each of us differ, at roughly 1/1000bp or 0.1%. Therefore using multiple DNA sources is not a major problem, indeed it provided many single nucleotide polymorphisms (SNPs) employed for mapping and evolutionary studies. 16. Our genome was already known to be around 3 Gbp, and indeed the draft sequences were both in the 2.9 Gbp range. Celera assembled theirs from their ±5X WGS, but added in the public sequences after shredding each BAC clone sequence into little bits. Even then the millions of transposons and other repeats caused problems. The public project at this point still had 75% of 33,000 BACs in draft, not finished sequence, and the assembly was a mess. Enter Jim Kent (below), a biocomputing graduate student at UC Santa Cruz, who recognized that the many available cDNAs could be used to orient the segments with exons they matched, and essentially single-handedly assembled the draft public genome sequence. “The human genome is a lot of work to sequence and put together, but it's not a human invention, and not something that we scientists have added so much value to. I feel most of the value is there from 3 billion years of evolution and I really think that it belongs to everybody." 17. Celera made their genome assembly available on DVD upon publication, but their website restricted downloads to 1Mbp per week. The public project was made available at the UCSC Human Genome Browser, as well as Ensembl in Europe and GenBank in the USA, free for downloading.
18. The UCSC Human Genome Browser. Here’s an example of a big gene, ITPR1, encoding inositol 1,4,5-triphosphate receptor, type 1. It has ±50 exons (vertical lines), sometimes alternatively spliced according to one of the cDNAs (U23850), with small to huge introns. The largest intron in this gene is around 110 kbp, and the gene is about 360 kbp. Compare that to the entire Mycoplasma genome at ±570 kbp. The largest human gene is dystrophin (DMD) at 2.4 Mbp, the gene mutated in Duchenne muscular dystrophy. The longest coding region is titin at ±81 kbp in 178 exons, one of which is the longest single exon of 17 kbp, encoding the longest protein at ±27,000 aa - titin is a “spring” that connects the Z and M lines in muscle fibers. There are also tiny single exon genes, and so many pseudogenes we still don’t have a final gene count.
19. We have not formally discussed how genes are modeled. There are broadly four kinds of evidence that can be used, each with advantages and disadvantages. • De novo modeling uses features of ORFs, such as biased AT/GC content, and the likely locations of intron donor (!GTaagt) and acceptor (pppppppprpAG!) sites (! is where the spliceosome cuts, and the GT and AG are absolutely required – except sometimes GC). Clearly this is going to be error-prone, with exons missed, genes truncated or fused, etc. • Evidence-based modeling is clearly one of the best approaches, using known cDNA or EST sequences to determine where exons/introns are and their boundaries. Unfortunately even this can be led astray with aberrant splicing, read-through of introns, etc. And it is limited by the coverage of the transcriptome, especially genes rarely expressed in a few cells, etc. • Comparative modeling uses the sequences of proteins from other organisms to align those to translated versions of the genome and divine the locations of exons and introns. For highly conserved proteins it works well in conjunction with de novo modeling to specific the intron boundaries, but is easily misled if an error was made in gene modeling in the other species. And it is nearly useless for rapidly evolving divergent proteins like my chemoreceptors. • We’ve seen earlier how evolutionary signatures (no frameshifting indels, third codon position changes, and conservative amino acid replacements) can be used to identify exons, but this only works when one has the genome sequences of several closely related species, so was not employed until recently. And it needs to be done along with de novo modeling. • In practice, gene annotation pipelines have been developed that simultaneously combine the top three and even the fourth method, although even these can seldom recognize pseudogenes.






















