Download

1 / 73
740 likes | 1.14k Views
4. Gene Expression Data Analysis. EECS 600: Systems Biology & Bioinformatics Instructor: Mehmet Koyuturk. Analyzing Gene Expression Data. Clustering How are genes related in terms of their expression under different conditions? Differential gene expression

E N D
4. Gene Expression Data Analysis EECS 600: Systems Biology & Bioinformatics Instructor: MehmetKoyuturk
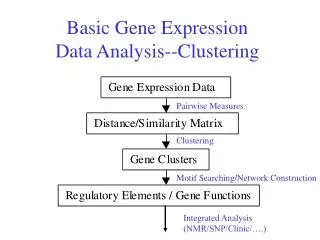
Analyzing Gene Expression Data • Clustering • How are genes related in terms of their expression under different conditions? • Differential gene expression • Which genes are affected by change in condition, tissue, disease? • Classification (supervised analysis) • Given expression profile for a gene, can we assign a function? • Given the expression levels of several genes in a sample, can we characterize the type of sample (e.g., cancerous or normal)? • Regulatory network inference • How do genes regulate each others expression to orchestrate cellular function? EECS 600: Systems Biology & Bioinformatics
Clustering • Group similar items together • Clustering genes based on their expression profiles • We can measure the expression of multiple genes in multiple samples • Genes that are functionally related should have similar expression profiles • Gene expression profile • A vector (or a point) in multi-dimensional space, where each dimension corresponds to a sample • Clustering of multi-dimensional real-valued data is a well-studied problem EECS 600: Systems Biology & Bioinformatics
Motivating Example Expression levels of 2,000 genes in 22 normal and 40 tumor colon tissues (Alon et al. , PNAS, 1999) EECS 600: Systems Biology & Bioinformatics
Applications of Clustering • Functional annotation • If a gene with unknown function is clustered together with genes that perform a particular function, then that is likely to be associated with that function • Identification of regulatory motifs • If a group of genes are co-regulated, then it is likely that their regulation is modulated by similar transcription factors, so looking for common elements in the neighborhood of the coding sequences of genes in a cluster, we can identify regulatory motifs and their location (promoters) • Modular analysis EECS 600: Systems Biology & Bioinformatics
Gene Expression Matrix n samples • Generally, m >> n • m = O(103) • n = O(101) • Each row is an n-dimensional vector • Expression profile m genes EECS 600: Systems Biology & Bioinformatics
Proximity Measures • How do we decide which genes are similar to each other? • Euclidian distance • Manhattan distance EECS 600: Systems Biology & Bioinformatics
Distance • Minkowski distance • General version of Euclidian, Manhattan etc. • p is a parameter EECS 600: Systems Biology & Bioinformatics
Normalization • If we want to measure the distance between directions rather than absolute magnitude, it may be necessary to standardize mean and variation of expression levels for each gene EECS 600: Systems Biology & Bioinformatics
Correlation • The similarity between the variation of two random variables • A vector is treated as sampling of a random variable • Covariance EECS 600: Systems Biology & Bioinformatics
Pearson Correlation Coefficient • Pearson correlation coefficient • Pearson correlation is equal to the cosine of the angle (or inner product of) normalized expression profiles • Pearson correlation is normalized EECS 600: Systems Biology & Bioinformatics
Euclidian Distance & Correlation • Euclidian distance (normalized) and Pearson correlation coefficient are closely related • These are the two most commonly used proximity measures in gene expression data analysis • Without loss of generality, we will use to denote the distance between two expression profiles EECS 600: Systems Biology & Bioinformatics
Other Measures of correlation • Pearson is vulnerable to outliers • If two genes have very high expression in a single profile, it might dominate to show that the two expression levels are highly correlated • Jackknife correlation: Estimate n correlations by taking each dimension (sample) out, take the minimum among them • Pearson is not robust for non-Gaussian distributions • Spearman’s rank order correlation coefficient: Rank expression levels, replace each expression level with its rank • More robust against outliers • A lot of loss of information EECS 600: Systems Biology & Bioinformatics
Clustering Methods • Hierarchical clustering • Group genes into a tree (a.k.a, dendrogram), so that each branch of the tree corresponds to a cluster • Higher branches correspond to coarser clusters • Partitioning • Partition genes into several groups so that similar genes will be in the same partition EECS 600: Systems Biology & Bioinformatics
Hierarchical clustering • Direction of clustering • Bottom-up (agglomerative): Start from individual genes, join them into groups until only one group is left • Top-down (divisive): Start with one group consisting of all genes, keep partitioning groups until each group contains exactly one gene • Agglomerative clustering is computationally less expensive • Why? • Hierarchical clustering methods are greedy • Once a decision is made, it cannot be undone EECS 600: Systems Biology & Bioinformatics
Agglomerative clustering • Start with m clusters: Each cluster contains one gene • At each step, choose two clusters that are closest (or most correlated), merge them • How do we evaluate the distance between two clusters? • Single-linkage: If clusters contain two very close genes, than the clusters are close to each other EECS 600: Systems Biology & Bioinformatics
Agglomerative Clustering • Complete linkage: Two clusters are close to each other only if all genes inside them are close to each other • Group average: Two clusters are close to each other if their centers are close to each other EECS 600: Systems Biology & Bioinformatics
Divisive Clustering • Recursive bipartitioning • Find an “optimal” partitioning of the genes into two clusters • Recursively work on each partition • Since the number of clusters is an issue for partitioning based clustering algorithms, the magic number 2 solves a lot of problems • May be computationally expensive • The problem is “global” • At every level of the tree, we have to work on all of the genes • If tree is imbalanced, there might be as many as m levels • With a reasonable stopping criterion, maybe considered a partition-based clustering as well EECS 600: Systems Biology & Bioinformatics
Partition Based Clustering • Find groups of genes such that genes in each group are similar to each other, while being somewhat less similar to those in other clusters • Easily interpratable • Especially, for large datasets (as compared to hierarchical) EECS 600: Systems Biology & Bioinformatics
Number of Clusters • Clustering is “unsupervised”, so generally we do not have prior knowledge on how many clusters underly the data • It is very difficult to partition data into an “unknown” number of clusters • Most algorithms assume that K (number of clusters) is known • Try different values of K, find the one that results in best clustering • Very expensive EECS 600: Systems Biology & Bioinformatics
Overlapping vs. Disjoint Clusters • Genes do not have a single function • Most genes might be involved in different processes, so their expression profiles might demonstrate similarities with different genes in different contexts • Can we allow a gene to be included in more than one cluster? • Allowing overlaps between clusters poses additional challenges • To what extent do we allow overlaps? (We definitely don’t want to identify two identical clusters) EECS 600: Systems Biology & Bioinformatics
Fuzzy Clustering • Assign weights to each gene-cluster pair, showing the extent (or likelihood) of the gene belonging to the cluster • Difficult interpretation • Partitioning is a special case of fuzzy clustering, where the weights are restricted to binary values • Hierarchical clustering is also “fuzzy” in some sense • Continuous relaxation might alleviate computational complexity as well EECS 600: Systems Biology & Bioinformatics
K-Means Clustering • The most famous clustering algorithm • Given K, find Kdisjoint clusters such that the total intracluster variation is minimized Cluster mean: Intracluster variation: Total intracluster variation: EECS 600: Systems Biology & Bioinformatics
K-Means Algorithm K-Means is an iterative algorithm that alters parameters based on each other’s values until no improvement is possible 1. Choose K expression profiles randomly, designate each of them as the center of one of the K clusters 2. Assign each gene to a cluster 2.1. Each gene is assigned to the cluster with closest center to its profile 3. Redetermine cluster centers 4. If any gene was moved, go back to Step 2, else stop EECS 600: Systems Biology & Bioinformatics
Sample Run of K-Means EECS 600: Systems Biology & Bioinformatics
Self Organizing Maps • Just like K-means, we have K clusters, but this time they are organized into a map • Often a 2D grid • We want to organize clusters so that similar clusters will be in proximity in the map • A way of visualizing in low-dimensional (2D) space • Just like K-means, each cluster is associated with a weight vector • It was the cluster center in K-means • Each weight vector is first initialized randomly to some gene’s expression profile EECS 600: Systems Biology & Bioinformatics
SOM Algorithm • At each step, a gene is selected at random • The distance between the gene’s expression profile and each cluster’s weight vector is calculated, and the cluster with closest weight vector becomes the winner • The winner’s and its neighbors’ (according to the 2D mapping) weight vectors are adjusted to represent the gene’s expression profile better • Cjis the winner cluster for gene i at time t • αis a decreasing function of time, θis the neighborhood function EECS 600: Systems Biology & Bioinformatics
Sample SOM Output EECS 600: Systems Biology & Bioinformatics
Gene Co-expression Network • Nodes represent genes • Weighted edges between nodes represent proximity (correlation) between genes’ expression profiles • This is indeed a way of predicting interactions between genes EECS 600: Systems Biology & Bioinformatics
Graph Theoretical Clustering • Partition the graph into heavy subgraphs • Maximize total weight (number of edges) inside a cluster • Minimize total weight (number of edges) between clusters • Heuristic algorithms • CLICK: Recursive min-cut • CAST: Iterative improvement one by one for each cluster • Loss of information? EECS 600: Systems Biology & Bioinformatics
Model Based Clustering • Generating model • Each cluster is associated with a distribution (that generates expression profiles for associated genes) specified by model parameters • The probability that a gene belongs to a cluster is specified by hidden parameters • Expectation Maximization (EM) algorithm • Start with a guess of model parameters • E-step: Compute expected values of hidden parameters based on model parameters • M-step: Based on hidden parameters, estimate model parameters to maximize the likelihood of observing the data at hand, iterate • K-means is a special case EECS 600: Systems Biology & Bioinformatics
Evaluation of Clusters • In general, we want to maximize intra-cluster similarity, while minimizing inter-cluster similarity • Homogeneity, separation • Based on the proximity metric • Reference partition • Information on “true clusters” that comes from a different source (apart from expression data) • Molecular annotation (e.g., Gene Ontology) • Jaccard coefficient, sensitivity, specificity • Cluster annotation • Processes that are significantly enriched in a cluster EECS 600: Systems Biology & Bioinformatics
Homogeneity & Separation • Heterogeneity (or homogeneity in reverse direction) • How similar are the genes in one cluster? • Separation • How dissimilar are different clusters? • Good clustering: high heterogeneity, low separation EECS 600: Systems Biology & Bioinformatics
Overall Quality • Overall heterogeneity • Overall separation • How do these change with respect to number of clusters? • Can we optimize these values to choose the best number of clusters? EECS 600: Systems Biology & Bioinformatics
Bayesian Information Criterion • A statistical criterion for evaluating a model • Penalizes model complexity (number of free parameters to be estimated) • k is the number of free parameters in the model, which increases with the number clusters • RSS is the “total error” in the model • Trade-off number of clusters and optimization function to choose the best number of clusters EECS 600: Systems Biology & Bioinformatics
Reference Partitioning • If there is information about “ground truth” from an independent source, we can compare our clustering to such reference partitioning • Pairwise assessment • Let Cij = 1 if gene i and gene j are assigned to the same cluster by the clustering algorithm, 0 otherwise • Let Rij = 1 if gene i and gene j are in the same cluster according to reference partition EECS 600: Systems Biology & Bioinformatics
Comparing Partitions • Rand index (symmetric) • Jaccard coefficient (sparse) • Minkowski measure (sparse) EECS 600: Systems Biology & Bioinformatics
Cluster Annotation • Clustering results in groups of genes that are co-expressed (or co-regulated) • For each group, can we tell something about the biological phenomena that underlies our observation (their co-expression)? • We have partial knowledge on the function of many individual genes • Gene Ontology, COG (Clusters of Ortholog Groups), PFAM (Protein Domain Families) • Taking a statistical approach, we can assign function to each group of genes • A function popular in a cluster is associated with that cluster EECS 600: Systems Biology & Bioinformatics
Gene Ontology • Ontology: Study of being (e.g., conceptualization) • Gene Ontology is an attempt to develop a standardized library of cellular function • Unified view of life: Processes, structures, and functions recur in diverse organisms • Three concepts of Gene Ontology • Biological process: A recognized series of events or molecular functions (e.g., cell cycle, development, metabolism) • Molecular function: What does a gene’s product do? (e.g., binding, enzyme activity, receptor activity) • Cellular component: Localization within the cell (e.g., membrane, nucleus, ubiquitin ligase complex) EECS 600: Systems Biology & Bioinformatics
Hierarchy in Gene Ontology • Gene Ontology is hierarchical • A process might have subprocesses • Seed maturation is part of seed development • A process might be described at different levels of detail • Seed dormation is a(n example of) seed maturation • Same for function and component • Gene Ontology terms are related to each other via “is a” and “part of” relationships • If process A is part of process B, then A is B’s child (B is A’s parent); B involves A • If function C is a function D, then C is D’s child; C is a more detailed specification of D EECS 600: Systems Biology & Bioinformatics
GO Hierarchy is a DAG • Gene Ontology is hierarchical, but the hierarcy is not represented by a tree, it is represented by a directed acyclic graph (DAG) • A GO term can have multiple parents (and obviously a GO term might (should?) have multiple children) EECS 600: Systems Biology & Bioinformatics
Annotation • GO-based annotation assigns GO terms to a gene • A gene might have multiple functions, can be involved in multiple processes • Multiple genes might be associated with the same function, multiple genes take part in a process • True-path rule • If a gene is annotated with a term, then it is also annotated by its parents (consequently, all ancestors) • How does the number of genes associated with each term changes as we go down on the GO DAG? EECS 600: Systems Biology & Bioinformatics
GO Annotation of Gene Clusters • There a |C| genes in a cluster C • |T| genes are associated with GO term t • |C ∩ T| genes are in C and are associated with t • What is the association between cluster C and term t? • If we chose random clusters, would we be able to observe that at least this many (|C ∩ T|) of the |C| genes in C are associated with t? • What is the probability of this observation? • Statistical significance based on hypergeometric distribution EECS 600: Systems Biology & Bioinformatics
Hypergeometric Distribution • We have n items, m of which are good • If we choose r items from the entire set of items at random, what is the probability that at least k of them will be good? • n is the number of genes in the organism • m=|T|, r=|C|, k= |C ∩ T| • The lower p is, the more likely that there is an underlying association between the term and the cluster (the term is significantly enriched in the cluster) EECS 600: Systems Biology & Bioinformatics
GO Hierarchy & Cluster Annotation • How specific (general) is the annotation we attach to a cluster? • If a cluster is larger, then it might correspond to a more general process • Some processes might be over-represented in the study set • How do we find the best location of a cluster in GO hierarchy? • Parent-child annotation • Condition probability of enrichment of a term in a cluster on the enrichment of its parent terms in the cluster • The gene space is defined as the set of genes that are associated with t’s parents EECS 600: Systems Biology & Bioinformatics
Parent-Child Annotation EECS 600: Systems Biology & Bioinformatics
Multiple Hypotheses Testing • The p-value for a single term provides an estimate of the probability of having the observed number of genes attached to that particular term • We have many terms, even if the likelihood of enrichment is small for a particular term, it might be very probable that one term will be enriched as much as observed in the cluster • We have to account for all hypotheses being tested simultaneously • Bonferroni correction: Apply union rule, add all p-values • Which terms should we consider while correcting for multiple hypotheses for a single term? EECS 600: Systems Biology & Bioinformatics
Representativity of Terms • How good does a significantly enriched term represent a cluster? • How many of the genes in the cluster are attached to the term? • How many of the genes attached to the term are in the cluster? • For term t that is significantly enriched in cluster C • Specificity: |C ∩ T|/|C|, a.k.a. precision • Specificity: |C ∩ T|/|T|, a.k.a. recall EECS 600: Systems Biology & Bioinformatics
Biclustering • A particular process might be active in certain conditions • A group of genes might be expressed (or up-regulated, supressed, co-regulated, etc.) in only a subset of samples • They might behave almost independently under other conditions EECS 600: Systems Biology & Bioinformatics