Download

1 / 38
380 likes | 561 Views
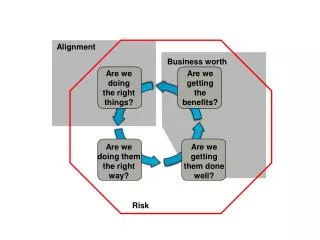
Alignment Problem. (Optimal) pairwise alignment consists of considering all possible alignments of two sequences and choosing the optimal one. Sub-optimal (heuristic) alignment algorithms are also very important: e.g. BLAST. Key Issues. Types of alignments (local vs. global)

E N D
Alignment Problem • (Optimal) pairwise alignment consists of considering all possible alignments of two sequences and choosing the optimal one. • Sub-optimal (heuristic) alignment algorithms are also very important: e.g. BLAST
Key Issues • Types of alignments (local vs. global) • The scoring system • The alignment algorithm • Measuring alignment significance
Types of Alignment • Global—sequences aligned from end-to-end. • Local—alignments may start in the middle of either sequence • Ungapped—no insertions or deletions are allowed • Other types: overlap alignments, repeated match alignments
Local vs. Global Pairwise Alignments • A global alignment includes all elements of the sequences and includes gaps. • A global alignment may or may not include "end gap" penalties. • Global alignments are better indicators of homology and take longer to compute. • A local alignment includes only subsequences, and sometimes is computed without gaps. • Local alignments can find shared domains in divergent proteins and are fast to compute
How do you compare alignments? • Scoring scheme • What events do we score? • Matches • Mismatches • Gaps • What scores will you give these events? • What assumptions are you making? • Score your alignment
Scoring Matrices • How do you determine scores? • What is out there already for your use? • DNA versus Amino Acids? • TTACGGAGCTTC • CTGAGATCC
Multiple Sequence Alignment Global versus Local Alignments • Progressive alignment • Estimate guide tree • Do pairwise alignment on subtrees ClustalX
Improvements • Consistency-based Algorithms • T-Coffee - consistency-based objective function to minimize potential errors • Generates pair-wise global (Clustal) • Local (Lalign) • Then combine, reweight, progressive alignment
Iterative Algorithms • Estimate draft progressive alignment (uncorrected distances) • Improved progressive (reestimate guide tree using Kimura 2-parameter) • Refinement - divide into 2 subtrees, estimate two profiles, then re-align 2 profiles • Continue refinement until convergence
Software • Clustal • T-Coffee • MUSCLE (limited models) • MAFFT (wide variety of models)
Comparisons • Speed • Muscle>MAFFT>CLUSTALW>T-COFFEE • Accuracy • MAFFT>Muscle>T-COFFEE>CLUSTALW • Lots more work to do here!
Modern Sequencing Methods • Sanger (1982) introduced a sequencing method amenable to automation. • Whole-genome sequencing: Clone-By-Clone vs. Shotgun Assembly • Drosophila melongaster sequenced (Myers et al. 2000) • Homo sapien sequenced (Venter et al. 2001)
Sanger (1982) introduced chain-termination sequencing. Main idea: Obtain fragments of all possible lengths, ending in A, C, T, G. Using gel electrophoresis, we can separate fragments of differing lengths, and then assemble them.
Automated Sequencing Perkin-Elmer 3700: Can sequence ~500bp with 98.5% accuracy
Reads and Contigs Sequencing machines are limited to about ~500-750bp, so we must break up DNA into short and long fragments, with reads on either end. Reads are then assembled into contigs, then scaffolds.
Clone-by-Clone vs. Shotgun • Traditionally, long fragments are mapped, and then assembled by finding a minimum tiling path. Then, shotgun assembly is used to sequence long fragments. • Shotgun assembly is cheaper, but requires more computational resources. • Drosophila was successfully sequenced using shotgun assembly.
Difficulties? • Good coverage does not guarantee that we can “see” repeats. • Read coverage is generally not “truly” random, due to complications in fragmentation and cloning. • Any automated approach requires extensive post-processing. • Phrapwww.phrap.org
The Fruit Fly • Drosophila melongaster was sequenced in 2000 using whole genome shotgun assembly. • Genome size is ~120Mbp for euchromatic (coding) portion, with roughly 13,600 genes. • The genome is still being refined.
NIH used a Clone-By-Clone strategy; Celera used shotgun assembly. Celera used 300 sequencing machines in parallel to obtain 175,000 reads per day. Efforts were combined, resulting in 8x coverage of the human genome; consensus sequence is 2.91 billion base pairs.
Abstraction • The basic question is: given a set of fragments from a long string, can we reconstruct the string? • What is the shortest common superstring of the given fragments?
Overlap-Layout-Consensus • Construct a (directed) overlap graph, where nodes represent reads and edges represent overlap. Paths are contigs in this graph. • Problem: Find the consensus sequence by finding a path that visits all nodes in layout graph. • Note: This is an idealization, since we must handle errors!
Approximation Algorithms • The shortest common superstring problem is NP-complete. • Greedily choosing edges is a 4-approximation, conjectured to be a 2-approximation. • Another idea: TSP has a 2-approximation if the edge weights are metric (Waterman et al. 1976 gives such metrics).
Handling Repeats • We can estimate how much coverage a given set of overlapping reads should yield, based on coverage. • Repeats will “seem” to have unusually good coverage. • Celera’s algorithms are proprietary, but there is no explicit way to handle repeats in the overlap-layout-consensus paradigm.
Hybridization Suppose we had a way to probe fragments of length k that were present in our sequence, from a hybridization assay. Commercial products: Affymetrix GeneChip, Agilent, Amersham, etc.
Sequencing-By-Hybridization • Then instead of reads, we have regularly sized fragments, k-mers. • Construct a multigraph G with (k-1)-mers as nodes, with edges representing k-mers. G is a de Bruijn graph. • Idea: An Eulerian path in G corresponds to the assembled sequence, and we don’t lose repeats (Pevzner 1989).
Bridges of Königsberg Theorem (Euler 1736): A graph has a path visiting every edge exactly once if and only if it is connected and has 2 or fewer vertices of odd degree.
Pros and Cons • An Eulerian path in a graph can be found in linear time, if one exists. • Errors in the hybridization experiments may prevent us from finding a solution. • Can we just use reads as “virtual” hybridization data?
Graph Preprocessing • Read errors mean up to k missing/erroneous edges. But we cannot correct this until we are done assembling! • Greedily mutate reads to minimize size of set of k-mers. • We also need to deal with repeats, which requires contracting certain paths to single edges…
Sequencing parameters • Difficulty and cost of large-scale sequencing projects depend on the following parameters: • Accuracy • How many errors are tolerated • Coverage • How many times the same region is sequenced • The two parameters are related • More coverage usually means higher accuracy • Accuracy is also dependent on the finishing effort
Sequence accuracy • Highly accurate sequences are needed for the following: • Diagnostics • e.g., Forensics, identifying disease alleles in a patient • Protein coding prediction • One insertion or deletion changes the reading frame • Lower accuracy sufficient for homology searches • Differences in sequence are tolerated by search programs
Sequence accuracy and sequencing cost • Level of accuracy determines cost of project • Increasing accuracy from one error in 100 to one error in 10,000 increases costs three to fivefold • Need to determine appropriate level of accuracy for each project • If reference sequence already exists, then a lower level of accuracy should suffice • Can find genes in genome, but not their position
Sequencing coverage • Coverage is the number of times the same region is sequenced • Ideally, one wants an equal number of sequences in each direction • To obtain accuracy of one error in 10,000 bases, one needs the following: • 10x coverage • Stringent finishing • Complete sequence • Base-perfect sequencing
NCBI Genome Summary • NCBI