Download

1 / 22
220 likes | 227 Views
This paper discusses the use of evolutionary algorithms to find the optimal gene sets for microarray prediction in distinguishing different cells, particularly in diseases like cancer.

E N D
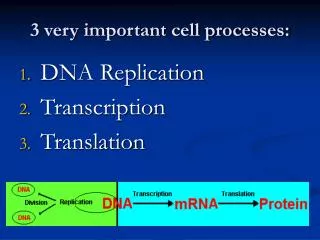
Evolutionary Algorithms for Finding Optimal Gene Sets in Micro array Prediction.J. M. DeutschPresented by: Shruti Sharma
Distinguishing one cell from other is very important and can be used in many diseases like cancer. cDNA and oligonucleotide microarray have been used successfully for the same purpose ie distinguishing one cell from another.
Approaches Used to Classify Microarray Data • Artificial Neural Network • Logistic Regression • Support Vector Machines • Coupled Two Way Clustering • Weighted Votes-Neighborhood Analysis • Feature Selection Technique
Deciding for the Predictor gene • Deciding for the Predictor gene is very important to classify the sample using microarray. • Too few genes • Too many genes.
Choosing of the optimal set of genes • Neighborhood Analysis • Principle Component Analysis • Gene Shaving
Gene selection : important part of prediction algorithm. • All high ranked genes are not chosen.
GESSES (Genetic Evolution of Sub Sets of Expressed Data) • Use replication algorithm used in quantum simulations and protein folding. • Consider the group of predictors and finds relevant genes. • It add and delete the genes continuously until an optimal performance has been achieved. • Blue cell tumor number of genes reduced from 96 to 15.Similarly good results were obtained for B-cell Lymphoma.
Overview of Algorithm • One which makes fewest mistakes on test data are the best predictors. • This algorithm uses a scoring scheme it gives higher scores when more data points are correctly classified. • LOOCV (leave –one-out cross validation) is used to calculate the score for a predictor. • The data is separated into clusters, each cluster corresponds to same type of cancer. • Wrapper Method- Search for the highest scoring predictor in the subset of the genes. • FilterMethod-filter gene pool and collects the most likely candidate gene
Terminology • Dt = {D1,D2…} Sample of microarray training data. • N- number of genes in the training data. • Gt - Complete set of genes(1 through N) • Gα- subset of complete gene set(α1, α2……. αm) • T - each sample D has classification of type T
Predictor • P - Predictor (a function that takes data D and output T) • K nearest neighbour search – to construct predictor. • Dt training data is compared with target data D by finding Euclidean distance between D and each vector in training set
Scoring Function • Iteratively single out one data point considering it to be pseudo test data. If the point is predicted correctly we add 1. • Consider shortest distance denoted by d1, d2….….dt • Take 2 shortest distance di and dj • Add C | di2 - dj2 | • C- constant chosen so that value of these added term <<<1 • Looping to entire data we calculate the total score.
Initial Gene Pool • To distinguish one gene from other we rank all training sets by expression levels. • Some genes give high level ( ti ) and some low levels ( tj ). • Some times they separate but many times they don’t separate clearly and overlap. • Those with overlaps are ranked low. • Thus we will choose the best M genes.
Replication Algorithm Suppose there is group of n gene subspaces ε ={G1,G2…..Gn} 1. For each G Єε produce a subspace as follows A. Set of genes G has genes {g1,g2……gm}. we randomly mutate the genes • ADD : choose a gene gr randomly and add it to G producing new set G’ of genes {g1,g2…gm, gr}. • DELETE : randomly delete a gene from G , new set will be with m-1 of total gene. • Keep G the same
Algorithm Continued B. Compute the difference in the score of the original gene G and mutated gene G’ δS=SG’-SG C. Compute weight for G’ w = exp(βδ S) β is inverse temperature 2. Z- Sum of these weights Normalize the weights by multiplying them by n/Z. 3. Replicate all the subspaces according to their weights w.
Annealing • As the system evolves the scoring function gives similar results. • To improve convergence temperature is made a function of spread in score. • The schedule for the temperature that worked the best was to lower the temperature with the fluctuation of the score from predictor to predictor.
Deterministic evolution • This is computationally expensive but performs better then the statistical method • The statistical method does not explore all combination of genes so can miss optimal gene combinations. • 1. Construct all distinct unions of the G’s in ensemble ε with individual gene gi in initial pool ie g1, g2, g3….gm, gi. • 2. Sort all these combinations by their score, keeping top n of them.
Small Round Blue Cell Tumors • Hard to classify by current techniques. • Hard to diagnose correctly as all appear similar. • Used 63 samples for training and tested with 20 using principle component analysis . • They reduced the number of genes to 96 yet classified data perfectly. • The same data set of 2308 genes was used and initial pool of genes was constructed by considering how well a gene discriminate cancer i from j. • For each combination top 10 genes best able to discriminate were selected • The Statistical Method was repeated .
Statistical Algorithm Using All Mutational Moves • Dimensions rises to maximum of about 16 • Wrong classification of predictors decreases from about 9 to 0.5 • By the end of the process the data was classified successfully. • At this point the temperature falls rapidly.
Deterministic Evolution method • Start with initial pool of 90 genes. • 15 overlapped, so total are 75. • n top =150 • Of the top 100 predictors all predicted the test data perfectly. • Average number of genes in predictor was 12.7. • Picture shows average number of genes in a predictor.
Leukemia • 2 types of leukemia 1. Acute Lymphoblastic Leukemia 2. Acute Myeloid Leukemia • From different cell types bone marrow samples were taken. • Each sample was analyzed by affymetrix microarrays having expression levels of 7129 genes • The data was divided into 38 training data points and 34 test points.
Statistical Algorithm Using All Mutational Moves • The average number of dimensions in a predictor rises to more than14 by iteration 63 • Dimensions declines by iteration 80 to dimension of only 2 • Average mistake remains constant at 1. • The methods predicts the training data perfectly
Conclusion • With GESSES we could distinguish Diffuse large B-cell lymphoma from b cell lymphoma. • Recently micro array data is used to distinguish 14 different kinds of tumors. • GESSES can help to practically use microarray data in cancer diagnosis and many other diseases.