Download

1 / 34
430 likes | 1.62k Views
NONMENDELİAN KALITIM. Prof.Dr.İlhan Sezgin TIBBİ GENETİK. NON-MENDELİAN KALITIM. A-Mendel kuralından sapmalar (değişken ifade –Variable expresitivite) a)Kompleks bir hastalığın özelliklerinde variasyonlar b)Hastalık şiddetinde variasyonlar

E N D
NONMENDELİAN KALITIM Prof.Dr.İlhan Sezgin TIBBİ GENETİK
NON-MENDELİAN KALITIM A-Mendel kuralından sapmalar (değişken ifade –Variable expresitivite) • a)Kompleks bir hastalığın özelliklerinde variasyonlar • b)Hastalık şiddetinde variasyonlar • c)İlerleme oranı ve başlangıç yaşındaki varyasyon • d)Monozigotlar arasındaki uyumsuzluk • e)Antispasyon • f)Mozaizm • g)Kimerizm
a. Kompleks bir fenotipin özelliginde varyasyonlar • Bir Mendel hastaltğının şiddeti yalnız ortaya çıkmadaki yetersizlik derecesi ile değil aynı zamanda bulunan spesifik özelliklerlede degişebilir. Böyle varyasyonlar birkaç sendrom veya farklı durum için incelenmiştir. Örneğin stickler sendromu (yada kalıtsal progressiv Arthroophtalmopaty) tip II yada tip XI kolejen yapıdaki degişmeler sonucunda oluşan bir bağ dokusu hastalığıdır. Stickler sendromlu bireyler miyopi, retinal detecment (retinanın damar tabakasından ayrılmasl) yarık damak ile birlikte giden küçük çene (Robin sequence), sağırlık, erken atritis, mitral kapak sarkması gibi çeşitli şekillerde kendini göstermektedir.
a. Kompleks bir fenotipin özelliginde varyasyonlar • Retinal ayrılma, sağırlık, Robin dizisi ve mitral sarkmasını kapsayan bulguların, hepsi aile bireylerinde birlikte görülmeyebilir • Bir başka örnek Waardenburg Sendromu tipidir ki bu sendromun özellikleri olan gözlerin, kulakların, deri ve saçların etkilendiği anormal pigmentasyon durumudurki pigment taşıyan hücrelerin anormal göçü sonucu oluşmaktadır. Bir aile içerisindeki bireyler derilerinde beyaz alanlar, beyaz saç perçemi, farklı renkte göz irisleri (heterochromia) yada bunlardan bazılarının birlikteliği şeklinde değişiklik gösterirlerki bunların bazıları senserionöral işitme kaybına sahip iken diğerleri değildir.
a. Kompleks bir fenotipin özelliginde varyasyonlar • Nörofibromatozis'te anormal bir pigmentasyon durumudur. Vucuttaki karekteristik kahverengi bölgeler cafe’ au lait alanlar mevcuttur. Etkilenmiş bireylerin vucutlarının herhangi bir yerinde bening tümörler gelişebilir. Bir aile içinde bazı bireylerde sadece cafe' au lait (sütlü kahve • şeklinde pigmentasyon) ve lischen nodulleri olarak adlandırılan gözlerde pigmentleşmiş bölgelerde bening tümörler varken, diğer aile bireylerinde deride tümörler gelişebilmektedir. Bireylerin %2'sinde malignant tümörler (nörofibromlar) gelişmektedir.
b. Hastalığın şiddetinde varyasyon • Aile bireyleri arasında bir hastalık bireyden bireye farklı şiddette ölümcül olabileceği gibi hafif geçirenlerde olabilir. • Örneğin Osteogenezis İmperfekta (cam kemik hastalığı) bazı çocuklar iskelet anomalileri ve kırıklarla doğarlar. Bunun aksine ebeveynlerinde belki hayat boyunca bir kırık olmamıştır. Belki sadece travma ile karşılaşınca fraktörler gelişmiş olabilir.
b. Hastalığın şiddetinde varyasyon • Marfan Sendromu bir bağ dokusu hastalığıdır. Bu sendromun hastada kendini gösterme şiddeti degişkenlik göstermektedir. Genellikle miyopi, lens luksasyonu (yer degiştirmiş lens) skolyaz, hiperekstansibl eklemler, mitral kapak prolapsusu, aort anevrizması spektrumu gösterir.
b. Hastalığın şiddetinde varyasyon Bir çok diğer iskelet metabolik sendromlarıda variable ekspresivite gostermektedir. Örneğin; • Diastrofik cücelik • Ehler- Danlos sendromu • Akandoplaziler • Metazifial ve Epizial Displaziler • Dyggvee -Melhior- Clausen Sendrumu • Muskopelisakkasidazlar.
c) İlerleme ve Başlangıç Yaşındaki Varyasyon • Ailede mevcut genetik hastalıklar hem başlama yaşı hemde ilerleme oranı bakımından etkilenmiş bireyler arasında degişkenlik göstermektedir. Polikistik böbrek hastalığı geçen zaman sürecinde ilerleyen otozomal dominant bir hastalıktırr. Bu hastalığın ilk belirtileri böbreklerde rastlanan kist gelişmeleridir. Zamanla kan damarları bozulmakta ve idrarda kan gelmesine sebep olmaktadtır. Çoğu polikistik böbrek hastalığı dializ ve transplantasyon gerektiren böbrekrek yetmezligine doğru ilerler. Doğal olarak kistler ve böbrek yetmezliği bir aile bireyinden diğerine ilerlemiş yaşlarda degişkenlik göstermektedir. Başlama yaşı,semptomların sayısı ve ilerleme oranındaki çeşitIiIik bu gib bazı hastalıklarda değişkenlik göstermektedir.
d.Monozigotik ikizler arasındaki uyumsuzluk • Variable ekspresivitenin özel bir durumu monozigotik ikizlerde görülmektedir. Monozigot ikizlerin aynı fenotipe sahip olmaları beklenir. Ancak her zaman öyle değildir. Bazı monozigotik ikizlerden biri normal erkek genitalyasına sahip iken diğerinde dişi genitalya tanımlanmıştır
d.Monozigotik ikizler arasındaki uyumsuzluk • Duchenne Muskular Distrofidede dişi monozigotik ikizlerin birinde buyük bir kas atrofisi ve hızlı ilerleme tanımlanırken diğerinde minimal ya da hiç kas atrofisi yoktur. Gerçekte orta derecede etkilenmiş ikizde hastalığın tek belirtisi sendromda kas enzimi keratin fosfokinaz konsantrasyonunun artmış olduğu görülmektedir.
d.Monozigotik ikizler arasındaki uyumsuzluk • Myotonic Distrofi (kasta kasma sorunu ile giden bir sendromdur) siirekli kas kosantraksiyonu (myotoni), katarakt, ~ dokUlmesi ve bazen mental gerilik'te gostermektedir. Myotonik ve kas gii~sUzlUgu problemi bireylerde y8§lanna gore fark etmektedir.{~kil-5)
ANTİCİPATİON • Miyotonik Distrofi gibi bazı otozomal dominant hastalıklar çocuklarda ebeveynlerden daha erken yaşlarda başlamakta ve daha ağır seyretmektedir. Bu tip kalıtıma "anticipation“ denmektedir.
ANTİCİPATİON • Miyotonik distrofi'de kaslar zayıflamış ve erimektedir. Önce yüz kaslarından başlar, bunun sonucu olarak pitoz gelişir ve göz kapakları düşer ve daha sonra boyun kasları, eller ve tüm vücuda yayılan genel bir miyotoni gelişir ve son aşamada kasların kasılıp gevşeme yetenekleri kaybolur. Miyotonik distrofide kalp kası ile düz kaslarda etkilenmektedir. Erken katarakt, immunoglobulin anomalileri ve sıklıkla hafif mental gerilik ilede birlikte gözlenir. Miyotonik distrofinin en önemli özelliklerden biride erişkin yaşlarda ortaya çıkmasıdır. Ancak bazı hasta ailelerin çocuklarında hastalık erken yaşlarda ortaya çıkmakta ve daha ağır seyretmektedir.
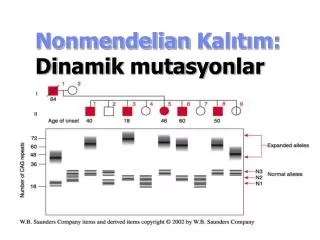
Myotonic Dystrophy CTG tekrarı • CTG tekrarı 3’UTR DMPK geni (19q13) – bir protein kinase • Normal allel – 5-30 CTG tekrar • Orta – 50-80 tekrar • Klasik allel – 80-150 tekrar • Kongenital – 2,000+ tekrar
Anticipation in Myotonic Dystrophy mild Repeat size in DMPK gene 60, 6 5, 7 classic classic 90, 5 96, 7 4, 12 congenital congenital 2150, 12 2900, 4
Sonuç • Üçlü nükleotid tekrarları hastalıklar oluşturabilir. • Myotonic Dystrophy • Huntington’s Disease • Fragile X Syndrome • Friedreich’s Ataxia
ANTİCİPATİON • Miyotonik geni klonlanıp incelendiği zaman genin 3' ucunda translokasyonu yapılamayan GCT ve CTG tripletlerinin bulunduğu görüldü. Normal şahıslarda bu tripletlerden 5-35 kopya bulunurken hasta şahıslarda 50' den fazla kopya bulunduğu ve kopya sayısının bazı hastalarda 100-1000 adete kadar çıktığı gözlenmiştir. Bu bölge stabil olmayan ve dinamik mutasyonlara açık bir bölgedir. Bu şunu göstermektedir. Miyotonik distrofideki ağırlıkla , erken yaşlarda ortaya çıkış ile tekrarlanan triplet sayısındaki artış arasında ilişki bulunmuştur .
ANTİCİPATİON • Konjenital myotonik distrofi yeni doganlarda gozlenir. Ve çok sayıda triplet tekrarı içerir. Nadir olarak gözlenmekle birlikte çok ciddi myotoni ve mental gerilik ile karekterizedir. Konjenital form her iki cinste ve hasta annelerin çocuklarında görülür.Çünkü en yüksek tekrarlar dişi mayozu sırasında meydana gelmektedir. Antisipanyonun varlığı miyotonik distrofiden başka ailevi parkison hastalığında, frajil- X sendromunda, ailevi şizofrenide, ailevi amilodotik nöropatide, kalıtsal nöro dejenaratif hastalıklarda, Spinoserebellar ataksi tip 2'de ,Huntington hastalığında ve goğüs kanserinde de ortaya konmuştur.
ANTİCİPATİON Antisipanyonun varlığı miyotonik distrofiden başka; • Ailevi parkison hastalığında • frajil- X sendromunda • Ailevi şizofrenide • Ailevi amilodotik nöropatide • Kalıtsal nöro dejenaratif hastalıklarda • Spinoserebellar ataksi tip 2'de • Huntington hastalığında • Göğüs kanserinde de ortaya konmuştur.
ANTİCİPATİON • Retinal aynlma, sagu-Itk, Robin dizisi ve mitral sarkmasw kapsayan bulgu1an, hepsi aile bireylerinde birlikte görülmeyebilir.
Mitokondriyal kalıtım • Hücre enerjisinin oluşumu Oksidatiffosporilasyon (OXPHOS) • Toksik reaktif oksijen türlerinin (ROS) OXPHOS sonucu oluşumu • Apopitozun başlangıcının düzenlenmesi
Mitokondrial DNA yapısında ağır (H, Guanin'ce zengin) ve hafif (L, Sitozince zengin) olmak üzere 2 iplik yer alır. H.iplik;12S ve 16S rRNA,12 polipeptit ve 14 tRNA kodlar. L.iplik; 1 polipeptit ve 8 tRNA kodlar.
mtDNA genom: • mtDNA : • tRNAs • rRNAs • cytochrome oxidase, NADH-dehydrogenase, & ATPase subunits. • Mitokondri genetikbilgisinin nukleusla ilgisi DNA: • DNA polymerase, replication factors • RNA polymerase, transcription factors • ribosomal proteins, translation factors, aa-tRNA synthetase • Additional cytochrome oxidase, NADH, ATPase subunits. • mtDNA genler her iki iplikte.
KODON Üniversal kod(nDNA) Mitokondrial kod(mtDNA) UGA STOP Trp AUA Ile Met CUA Leu Leu AGA Arg STOP AGG Arg STOP Üniversal kod ve mitokondrial kod arasındaki farklar.
Maternite analizi • Pilogenetik sistematik • Populasyon genetiği (ve conservation genetiği) • Forensics (maternal ID)
Mutasyonlar mtDNA ‘da hastalıklara yol açar: • 100 farklı mtDNA düzenlenmasi ve 50 farklı nokta mutasyonu bulunmuştur. • 1 / 8,500 mitokondriyal hastalığa rastlanır. • Missense mutasyonlar • Nokta mutasyonlar tRNA veya rRNA ‘da • Delesyon ve duplikasyonlar mtDNA’da
Mitokondriler maternal kalıtım gösterirler: • Memeli yumurtası 100,000 mitokondri DNAsı taşır. Mammalian sperm contain about 100 mtDNA copies
Mitokondrial Hastalıklar • Leber’in Herediter Optik Nöropatisi • Kronik Progressiv Eksternal Oftalmopleji • Kearn-Sayre Sendromu • Pearson Sendromu • Mitokondrial Ansefalomyopati,Laktik Asidoz,Stroke- Like Epizod • Miyoklonik Epilepsi ile (Ragged Red Fİbers) • Nöropati,Ataksi ve Retinitis Pigmentoza • Leigh Sendromu • Mitokondrial Sağırlık
İnsan mt hastalıklarına örnekler: • Leber’s hereditary optic neuropathy (LHON), OMIM-535000 • Körlük, optik sinir degenerasyonu • Mutasyonlarin ND1, ND2, ND4, ND5, ND6, cyt b, CO I, CO II, ve ATPase 6 elektron transport zincirini engellerler. • Kearns-Sayre Syndrome, OMIM-530000 • Göz kaslarında felç, pigment birikimi ve degenerasyon, ve kalp hastalıkları. • Delesyon mtDNA tRNAs. • Myoclonic epilepsy & ragged-red fiber disease (MERRF), OMIM-545000 • Spasm ve anormal dokular, kanda laktik asit birikimi, düzensiz hareketler • Nucleotid sustitüsyonu mtDNA lisine tRNA’da. • mtDNA hastalarve mutant mtDNA karışımı içerirler, hastalığın derecesi farklıdır