Download
1 / 31
330 likes | 527 Views
Nonmendelian Kalıtım : D i nami k muta sy on lar. Objecti fler. Üçlü nukleotid tekrarları F ragil X s e ndrom u , F riedreich’s ataxia, H untington’s hastalığı ve M yotonic d i stro fi. Tarihçe. 1991 ’de gözlendiler Üçlü nukleotidlerde stabil olmayan artışlar izlendi

E N D
Objectifler • Üçlü nukleotid tekrarları • Fragil X sendromu, Friedreich’s ataxia, Huntington’s hastalığıveMyotonic distrofi
Tarihçe • 1991’de gözlendiler • Üçlü nukleotidlerde stabil olmayan artışlar izlendi • Tekrarlar normalde populasyonda polimorfik, fakat ailelerde stabil • Tekrarlar etkilenen ailelerde stabildeğil • Artış çok farklı değerlerde olabilir
AUG TAA CGG CAG CTG GAA Myotonik Distrofi Fredreich’s Ataxia Fragil X Sendromu Fragil XE MR Spinobulbar Muscular Atrophy Huntington’s Disease Dentatorubral-Pallidoluyslan Atrophy Spinocerebellar Ataxia Type 1 Spinocerebellar Ataxia Type 1 Spinocerebellar Ataxia Type 2 Spinocerebellar Ataxia Type 6 Spinocerebellar Ataxia Type 7 Spinocerebellar Ataxia Type 8 Spinocerebellar Ataxia Type 12 Machado-Joseph Disease (SCA3) 14 stabil olmayan Trinukleotid tekrarı ve bunlarla ilgili hastalıklar
AUG TAA CGG CAG CTG GAA Myotonic Dystrophy Fredreich’s Ataxia Fragile X Syndrome Fragile XE MR Spinobulbar Muscular Atrophy Huntington’s Disease Dentatorubral-Pallidoluyslan Atropphy Spinocerebellar Ataxia Type 1 Spinocerebellar Ataxia Type 1 Spinocerebellar Ataxia Type 2 Spinocerebellar Ataxia Type 6 Spinocerebellar Ataxia Type 7 Spinocerebellar Ataxia Type 8 Spinocerebellar Ataxia Type 12 Machado-Joseph Disease (SCA3)
Intronik 3’ UTR 5’ UTR Polyglutamine Kodlama yapan bölgede • Ortak özellikler: • Nörolojik hastalık • Genelde Otozomal Dominant –X-bağlıveya resesif olabilirler • Azalmış penetransgösterebilirler AUG TAA CGG CAG CTG GAA
Slipped Mispairing underlies triplet repeat expansion Lagging Strand Template 5’ (A) Leading Strand Template 3’ 1 3 2 5’ 5’ (B) Okazaki Fragments Polymerization proceeds from 5’ to 3’ for the newly synthesized DNA strands. On the lagging strand, synthesis proceeds 5’ to 3’ for each Okazaki fragment, but overall lagging strand synthesis proceeds 3’ to 5’ as the fragments extend, meet and are ligated together, indicated by 1,2,3. 3’ 5’
Slipped Mispairing underlies triplet repeat expansion Lagging Strand Template 5’ Leading Strand Template (A) 3’ CGG CGG CGG 1 3 2 5’ 5’ (B) Okazaki Fragments Polymerization proceeds from 5’ to 3’ for the newly synthesized DNA strands. On the lagging strand, synthesis proceeds 5’ to 3’ for each Okazaki fragment, but overall lagging strand synthesis proceeds 3’ to 5’ as the fragments extend, meet and are ligated together, indicated by 1,2,3. 3’ 5’
Fragil X • X-bağlı dominant penetrans azlığı gösterir • Orta düzeyde mental retardasyon – 1/4000 erkeklerde; 1/8000 dişilerde • X kromozomunda (Xq27.3) “fragil” bölge dekondanse
(CGG)n A U G T A A 6 1 4 a m i n o a c i d s ( 6 9 k D ) 1 2 3 4 5 6 7 8 9 1 0 1 1 1 2 1 3 1 4 1 5 1 6 1 7 3 8 k b Fragil X Mental Retardasyon gen 1 FMR1
Klinik özellikler: Fragil X • Uzun yüz – • Büyük kulaklar • Mental gerilik (I.Q. 20-60) • Dikkat kusuru/hiperaktivite
2 yrs. 5 yrs. 22 yrs. Fragile X in males
Fragil X’de CGG 5’ UTR de FMR1 geninde • Allel tipleri: • Normal allel – 7-40 CGG • Premutasyon allel - 60-200 • Hasta allel - >200 den binlerce tekrara kadar • İşlev kaybı
Male Female 1/25 1/16 1/1000 1/350 1/4000 1/8000 Fragil X Transcription CpG Island Translation (CGG) n 3’ 5’ 40 repeats Common 41-60 repeats Intermediate Premutation 61-200 repeats > 200 repeats, methylated Full mutation
FMR1 Repeat Instability mean I 88 85,25 94,25 90 86,25 104 II 111 95 118 93 III >230 500 31 Full mutation Premutation Normal 32P-CGG probe of PCR
Functional domains in the FMR protein NLS KH1 KH2 NES RGG 111-152 206-280 281-422 425-441 527-552 111-152 206-280 281-422 425-441 527-552 Cytoplasmic protein NLS-nuclear localization sequence (Eberhardt, et al. 1996. Hum.Mol.Gen) KH-homology to hnRNP K (Siomi, et al. 1993. Cell) NES-nuclear export sequence (Eberhardt, et al. 1996. Hum.Mol.Gen) RGG-arginine-glycine-rich region (Siomi, et al. 1993. Cell)
FMRP mGluR Glu Spine maturation; synaptic plasticity Model for FMRP function in neurons AXON DENTRITE
Friedreich Ataxia 1/50,000 • otosomal recessive spinocerebellar ataxia www.barnstormers.org.uk/ images/jamie1.jpg
Friedreich Ataxia • AAG tekrarları intron frataxin de • AlelTipleri: • Normal allel - <34 tekrar • Taşıyıcı allel – 36-100 tekrar • Hasta allel - >100 tekrar AUG TAA CGG CAG CTG GAA
Frataxin mitokondrial protein Demir metabolismasında iş görür.
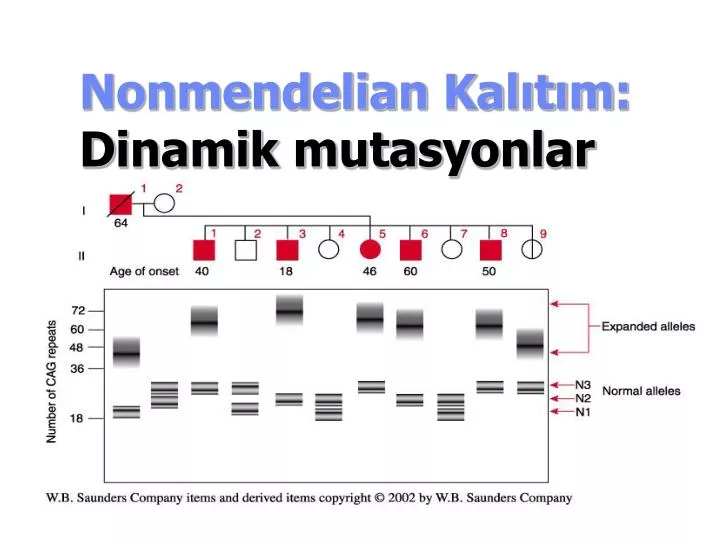
Huntington Disease (HD) • Otosomal Dominant (~1/25,000,) • Orta yaşta başlar ölümle sonuçlanır.
HD CAG tekrarı ile oluşur. • polyglutamin-kodlayan gende CAG ekson 1de • Normalde – 9-35 CAG tekrar • Hastalarda – 36+ tekrar üstü120 ye kadar.
Solid line – avg age at onset; shaded area shows rage of age of onset
Myotonic Dystrophy CTG tekrarı • CTG tekrarı 3’UTR DMPK geni (19q13) – bir protein kinase • Normal allel – 5-30 CTG tekrar • Orta – 50-80 tekrar • Klasik allel – 80-150 tekrar • Kongenital – 2,000+ tekrar
Anticipation in Myotonic Dystrophy mild Repeat size in DMPK gene 60, 6 5, 7 classic classic 90, 5 96, 7 4, 12 congenital congenital 2150, 12 2900, 4
Sonuç • Üçlü nükleotid tekrarları hastalıklar oluşturabilir. • Myotonic Dystrophy • Huntington’s Disease • Fragile X Syndrome • Friedreich’s Ataxia










