Download

1 / 20
200 likes | 324 Views
Bioinformatic discovery of microRNA precursors from human ESTs and introns. Sung-Chou Li Chao-Yu Pan Wen chang Lin 2006. Outline. Introduction Scanning method Result Specificity assessment Discussion Conclusion Reference. Introduction. Intron

E N D
Bioinformatic discovery of microRNA precursors from human ESTs and introns Sung-Chou Li Chao-Yu Pan Wenchang Lin 2006
Outline • Introduction • Scanning method • Result • Specificity assessment • Discussion • Conclusion • Reference
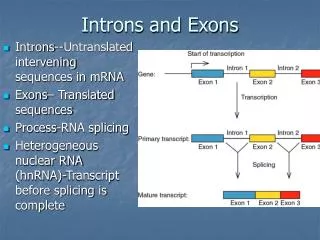
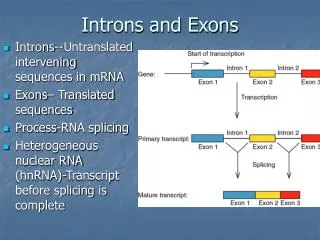
Introduction • Intron • DNA region that is not translated into protein • Consist by pre-mRNA and other RNAs • Remove by splicing • Containing 60 known pre-miRNA (release 5)
Introduction • Expressed Sequence Tag(EST) • Used to identify gene transcript, gene discovery and gene sequence determination • Containing 26 known pre-miRNA (release 5)
Introduction • microRNA (miRNA) • Non-protein-coding RNA • down-regulate gene expression • disease progression
Scanning Method • Direct-cloning • Traditional biological method in miRNA discovery • Limited by need of RNA start material • Few highly express miRNA constitute cloned product • miRNAs are unstable, easy to degraded
Scanning Method • Bioinformatic discovery Srnaloop Sequence & Structural RefSeqfilter Conservation examination
Srnaloop • Srnaloop is a BLAST-like algorithm that looks for short complementary words within a specified distance. • Uses dynamic programming to determine a complete alignment. Compared to BLAST, srnaloop supports shorter word lengths and aligns complementary base pairs. • Acquire 1,350,168 candidate hairpins( 359,360 from ESTs, 990,808 from introns).
Sequence & Structural features filter • GC contentDifferent types of genome sequence have different ranges of GC content, and pre-miRNAs are also expected to be so. • Core mfe and hairpin mfe(Minimum Free Energy)Hairpin structure results from intra-molecular base pairing with hydrogen bonding. Different pairing patterns result in distinctive stabilities and minimum free energies.Greater a number of paired bases within a hairpin implies greater stability and lower mfe
Sequence & Structural features filter • Ch_ratio core mfe / hairpin mfe, ch_ratio has a fixed distribution range • Acquire 113,484 candidate hairpins from introns and 67,215 from ESTs.
RefSeq filter • The Reference Sequence (RefSeq) database is a non-redundant collection of richly annotated DNA, RNA, and protein sequences. • RefSeq biological sequences are derived from GenBank records but differ in that each RefSeq is a synthesis of information. • NM access number are the tag to extract mRNA protein-coding sequence, if candidate matching these sequence will be removed. • Acquire 66,109 candidate from ESTs and 113,124 from introns.
Conservation Examination • miRNAs have been conserved among phylogenetically close species during evolution. • Searched putative pre-miRNAs against other published mammalian genomic sequences, namely mouse , rat and dog. • Conserved hairpin definition: Putative miRNAs of a hairpin has a contiguous >= 20-nt fragment that is identical to a subject sequence.
Conservation Examination HMDR are the pre-miRNA candidates conserved inall four genomes (human, mouse, dog and rat). After applying the final conservation filters, there were 208 qualified candidate hairpins in the HMDR set, 52 of which were known pre-miRNAs, resulting in 60.5% (52/86) sensitivity and 25.0% (52/208) specificity.
Specificity assessment(1) Using the 130 newly updated (release 8) pre-miRNA sequences as our validation test dataset. Detected 116 of the 130 input pre-miRNAs after the initial hairpin finding procedure. And then tested the sensitivity of the Sequence & Structural features filter led to 85% sensitivity. (previous sensitivity 86.5%)
Specificity assessment(2) This procedure is based on the fact that the fraction of miRNA encoding sequences in the human genome is very small; therefore, randomly extracted sequences are extremely unlikely to code for miRNA. randomly extracted 99,600 sequence fragments from intronic sequences (33,200 fragments), ESTs (33,200 fragments) and genomic sequences (33,200 fragments). The 332 known pre-miRNA sequences and the 99, 600 random sequences of 11 Mbps were applied to our discovery pipeline under the same hairpin finding parameters.
Specificity assessment(2) ESTs introns Genome Seq Scanning method 33200 11M 5 output (2,1,2) 1440M 33200 33200 332 known pre-miRNA sequences Scanning method 210 output
Specificity assessment(2) Of the 332 known pre-miRNAs, 210 survived the discovery pipeline as a true positive prediction value. 5 false positives in three independent experiments (2, 1 and 2 predicted candidates respectively), corresponding to an average of 1.67 false positives in 11 Mbps. Thus, because the initial are about 1,440 Mbps in length, we could theoretically generate 212 false positive candidates from similar size dataset. 210 (TP)/(210 (TP) + 212 (FP)) , The specificity is 49.7% by calculating the percentage ratio of where TP denotes true positives and FP denotes false positives.
Discussion Using intronic and EST sequences as raw data. Advantage: most of the ESTs and introns are well annotated, making it easy to acquire information associated with their expression patterns and levels. Disadvantage: all of the 207 known pre-miRNAs should have matches when searching against the human genome; in our data, however, only 60 and 26 pre-miRNAs matched to introns and ESTs, respectively, implying only 41.5% [(60 + 26)/207] coverage.
Conclusion This paper developed a new scanning method using criteria based on the features of 207 known pre-miRNAs to predict miRNAs from expressed sequences (ESTs and introns). And it achieve good sensitivity and specificity compared with other publishedworks.
References • Lai EC, Tomancak P, Williams RW, Rubin GM: Computational identification of Drosophila microRNA genes. Genome Biol2003 • Grad Y, Aach J, Hayes GD, Reinhart BJ, Church GM, Ruvkun G, Kim J: Computational and experimental identification of C. elegansmicroRNAs. Mol Cell 2003 • Lee Y, Tsai J, Sunkara S, Karamycheva S, Pertea G, Sultana R, Antonescu V, Chan A, Cheung F, Quackenbush J: The TIGR Gene Indices: clustering and assembling EST and known genes and integration with eukaryotic genomes.