Download

1 / 72
740 likes | 967 Views
Cystic Fibrosis. Dr Barry Linnane Paediatric Respiratory Consultant Mid-Western Regional Hospital Limerick. Introduction.

E N D
Cystic Fibrosis Dr Barry Linnane Paediatric Respiratory Consultant Mid-Western Regional Hospital Limerick
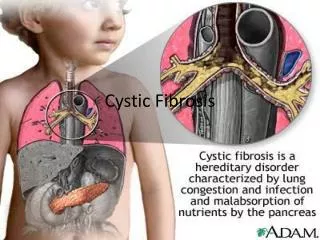
Introduction • It is customary to start any discussion regarding cystic fibrosis (CF) by pointing out that it is the most common life-shortening genetic condition affecting the Caucasian population (Littlewood, in Cystic Fibrosis, 2007. p. 3 – 19) • It is estimated there are approximately 60,000 affected individuals worldwide(Anselmo et al, in Pediatric Respiratory Medicine, 2008. p. 845 – 857) • The prevalence of the disease varies amongst populations, but the average birth prevalence in Caucasian populations is likely to be approximately 1 in 2,500 births, with a CF carrier frequency of 1 in 25 people (Walters et al, in Cystic Fibrosis, 2007 p. 21 – 45) • There are approximately 1,200 individuals with CF in Ireland (The Cystic Fibrosis Registry of Ireland Annual Report 2007) • There are 30-40 infants born each year with CF in Ireland. The birth prevalence is approximately 1 in 1460, with a CF carrier frequency of 1 in 19 (Wallis C, in Kendig’s Disorders of the Respiratory Tract in Children, 2006, p866 – 872)
Definition • There are numerous definitions of CF, all of which attempt to capture the essence of this complex disease, in a few succinct words. Dr Colin Wallis recently proposed the following definition: “A possible definition for the disease CF could involve the following sequence of pathologic events: • Disease usually arises from two disease-causing mutations in the gene encoding CFTR • Mutations result in changes to the fluid and electrolytes on cell surfaces • Changes may lead to abnormal secretions and inflammatory responses • These manifestations may predispose to obstruction and infection • Obstruction and infection may produce end-organ disease in tubular structures such as the upper and lower airways, vas deferens, gut, liver, and pancreas, with secondary impact on growth and nutrition.” (Wallis C, in Kendig’s Disorders of the Respiratory Tract in Children, 2006, p866 – 872)
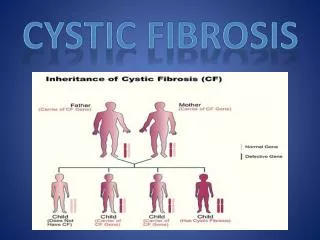
Genetics • CF is an autosomal recessive condition • In the late 1980s technical breakthroughs in gene analysis involving chromosome walking and jumping, and linkage disequilibrium analysis resulted in 1989 in the identification of the CF gene on the long arm of Chromosome 7 at the 7q31 locus (Kerem et al, 1989;245:1073) (Rommens et al, 1989;245:1059) (Riordan et al, 1989;245:1066) • As per convention the gene takes on the name of the protein it codes for, in this case, the cystic fibrosis transmembrane regulator CFTR (more about that later) • Over 1500 mutations of the CFTR gene have been identified (www.genet.sickkids.on.ca/cftr/) • However, deltaF508 is the dominant mutation seen in over 70% of CF alleles in the Caucasian population worldwide
Genetics • All the other mutations are individually rare, with only 11 found in more than 100 patients • 94% of the Irish CF population carries the deltaF508 mutation on at least one chromosome, with 64% being homozygous for the mutation (The Cystic Fibrosis Registry of Ireland Annual Report 2007) • The six next most common mutations are (The Cystic Fibrosis Registry of Ireland Annual Report 2007): • G551D • R117H • R560T/K • G542X • 1717-1 G-A • 621+1 G-T
5 major classes of CF mutations • Extensive research into genotype-phenotype correlations, along with knowledge of the molecular mechanisms by which genetic mutations result in defective proteins, has led to a classification system for CF mutations (Welsh, et al, 1993;73:1251) • It should be emphasized though, that most CFTR mutations result in the classical severe form of the disease • Class I: defective protein synthesis (e.g. G542X, 1717-1 G-A, 621+1 G-T) • Class II: defective protein processing (e.g. DF508) • Class III: defective regulation (e.g. G551D, R560T/K) • Class IV: defective conductance (e.g. (R117H) • Class V: reduced protein levels at the membrane
CFTR • The CFTR protein is a chloride channel at the apical membrane of epithelial cells (Riordan et al, 1989;245:1066) • It comprises of 1480 amino acids, and is part of the family of ATP-binding cassette (ABC) transporter proteins • Phosphorylation by protein kinase, under the control of cAMP, and hydrolysis of ATP, are essential for activating the chloride channel
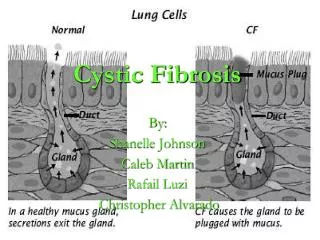
Normal mucociliary clearance • To understand the abnormality that occurs in CF we must first understand what is normal • The respiratory tract is lined by ciliated columnar epithelium • Resting on top of the epithelium is the Airway Surface Liquid (ASL) which is composed of an outer (towards the lumen) mucous layer, and an inner watery Peri-Ciliary Layer (PCL) which bathes the cilia (Matsui, et al, 1998;102:1125) • The PCL is approximately 7 microns deep, which is the length of an outstretched cilium • The arrangement is part of the innate immune system of the respiratory tract, and works by trapping bacteria in the mucous layer which is then transported in a cephalic direction by the mucociliary elevator to be coughed and swallowed or expectorated – a process known as mucociliary clearance
CF is a disease of the airways From the nose to the bronchioles the airway is lined by pseudo-stratified columnar epithelium
Peri-ciliary layer Airway lumen Airway surface liquid Mucus layer Epithelium Basement membrane
Cl- Na+ CFTR ENaC Ion Pump
Mucous layer Cl- Na+ 7 microns CFTR Peri-ciliary layer H2O H2O
Mucous layer 7 microns CFTR Peri-ciliary layer Cl- Na+ H2O H2O
Purulent tenacious mucous layer 3 microns CFTR Peri-ciliary layer Cl- Na+ H2O H2O
Normal mucociliary clearance • The volume of the ASL is tightly regulated by an active process of ion transport • Absorption of ASL: The epithelial Na channel (ENaC) actively transports Na from the ASL into the epithelial cell, with Cl following the electrochemical gradient, and H2O following the osmotic gradient • Secretion of ASL: In contrast CFTR actively transports Cl out of the epithelial cell into the ASL, with Na and H2O following passively • Therefore, if ASL volume is excessive, Na absorption increases, and as ASL reaches a physiological optimum Na absorption reduces, or stops. Cl secretion can then dominate if ASL volume is diminished.
CFTR in CF • In CF, derangement of CFTR in airway epithelium results in defective epithelial ion transport by two mechanism • Lack of functioning CFTR appears to release ENaC from tonic inhibition, which results in greater Na absorption, with Cl and H2O following, resulting in diminished ASL volume • Defective CFTR is unable to secrete Cl, which would have resulted in Na and H2O following, to rehydrate the ASL • The result of both of this processes is a reduction in the volume of the ASL, with the PCL dropping to a depth of 3 microns • As a consequence mucociliary clearance is deranged, and mucous plaques adhere to the epithelium • From this apparently innocuous start come all the devastating consequences of CF lung disease
Pathogenesis of airway disease • Mucociliary clearance is inhibited, as described above • The mucous is thick, and prevents penetration by immune cells (but not motile Pseudomonas aeruginosa) • The unregulated ENaC channels consume increased energy, and oxygen, resulting in a relatively hypoxic environment in the mucous plaques • The resultant micro-environment appears to specifically select for the typical CF airway pathogens, most notably P. aeruginosa • The presence of airway pathogens initiates a marked immune response lead by neutrophils. The massive neutrophil response results in the production of neutrophil elastase at levels that overwhelm the innate anti-proteases (e.g. alpha1 anti-trypsin)
Pathogenesis of airway disease • The resulting free neutrophil elastase degrades the structural proteins of the airway culminating in irreversible airway and parenchymal destruction • The problem is exacerbated by three further processes • Bacterial infection, and the resulting inflammation, induce increased mucin production • DNA from necrotic neutrophils and bacteria increase the tenacity of airway mucous • P. aeruginosa adapts to its new environment by producing biofilms which significantly reduce its susceptibility to eradication by antibiotics
Pancreatic disease • The pancreatic acinus secretes isotonic liquid, which is then modified in the pancreatic duct, to produce a fluid rich in HCO3- (Steward, et al, 2005;67:377) • Basolateral ion exchange mechanisms increase in the intracellular HCO3- concentration, with CFTR playing a role in transporting HCO3- into the lumin (Steward, et al, 2005;67:377) • CFTR appears to act as a conduit for anions other than Cl-, in particular HCO3-, which is transported at about 40% efficiency when compared with Cl- (Steward, et al, 2005;67:377) • In CF there is deficient secretion of HCO3-, and liquid, with resultant abnormally acidic and viscous ductal secretions, which likely lead to early activation of proteolytic enzymes within the gland, resulting in inflammation and destruction (Johansen, et al, 1968;1:455) (Kopelman, et al, 1985;312:329)
Sweat gland • The observation that sweat electrolytes are abnormal in CF represents on the seminal discoveries in CF (Di Sant'Agnese 1953;12:549) • Normally, isotonic sweat is produced in the sweat acinus, with Cl and Na being reabsorbed in the resorptive duct, resulting in hypotonic (Cl 10 – 50 mmol/L) sweat reaching the skin surface • In the CF sweat gland, isotonic sweat is produced in the acinus, but the CFTR dependent resorption of Cl is deficient resulting in abnormally high sweat Cl levels ( 60 – 120 mmol/L) reaching the skin surface • This forms the basis of the pilocarpine iontophoresis induction of sweating as part of the sweat test, which allows sufficient quantity of sweat for Cl and Na analysis, and hence the diagnosis of CF (Gibson, Cooke, 1959;23:545)
The genetics and pathology of CF come together when we focus on diagnosis
Diagnosis • The diagnosis of CF is usually made when an individual presents with suggestive clinical features such as recurrent chest infections, and evidence of malabsorption with failure to thrive and steatorrhoea; which is then confirmed on sweat testing with an elevated sweat chloride > 60 mmol/L. • 18 % present with Meconium ileus in the neonatal period • 15% present with a family history typically of an affected sibling
Clinical manifestations Or CF history in a sibling Or positive newborn screening Positive sweat test Or positive nasal potential difference Or two disease causing mutations identified In 1998 the CFF issued a consensus statement on the diagnosis of CF Plus
Clinical manifestations Or CF history in a sibling Or positive newborn screening Positive sweat test Or positive nasal potential difference Or two disease causing mutations identified In 1998 the CFF issued a consensus statement on the diagnosis of CF Plus
Symptoms at diagnosis (The Cystic Fibrosis Registry of Ireland Annual Report 2007)
Sweat Test • The sweat test was first described by Gibson and Cooke in 1959 and forms the gold standard for the diagnosis of CF (Gibson, Cooke,1959;23:545) • Guidelines for sweat testing have been published, and its is imperative they are closely followed in a laboratory with experience in sweat testing (Baumer, 2003;88:1126) • The technique uses iontophoresis to drive pilocarpine into the skin to induce local sweating • The sweat sample is collected either using the macroduct system or on filter paper • A minimum sample of 15 microL is required using the macroduct system, and of 75 mg when using filter paper
Sweat Test • Sweat Cl > 60 mmol/l is diagnostic of CF • Sweat Cl < 40 mmol/l rules out CF • Sweat Cl 40 – 60 mmol/l is equivocal • Sweat Na should not be used to diagnose CF because values of up to 80 mmol/l can be seen in conditions other than CF • However the sweat Na can be used to check the validity of the test as Na and Cl levels should be within 15 mmol/l of each other • Sweat Cl > 160 mmol/l are physiologically impossible and if detected require the sweat test to be repeated • Sweat conductivity should not be used
Respiratory • The fundamental problem in the CF airway is the build up of tenacious secretions resulting in airway occlusion, infection, inflammation, airway and lung parenchyma destruction, and bronchiectasis • The central components of CF respiratory care are: • Airway clearance • Antibiotic therapy
Airway infection • The term airway “colonization” has largely been abandoned when discussing airway pathogens, in favour of the term airway infection, recognising that the presence of pathogens in the lower airway is abnormal and associated with an inflammatory response. • Repeated airway infection leads to sustained inflammation which results in time in irreversible airway and lung parenchyma destruction • Respiratory failure remains the principal cause of premature death in patients with CF • Much of the ongoing care of patients therefore is focused on managing airway infection • It has been demonstrated that lower infection begins early in life, indeed in infancy • S. aureus and H. influenza are the typical initial infecting organisms, followed later by P. aeruginosa
Cystic Fibrosis Foundation Patient Registry Annual Data Report 2005
S. aureus • S. aureus is the most common organism infecting young children with CF (Elborn, et al, 1999;54:377), and remains an important pathogen throughout life, often present as a co-infection • There remains some debate regarding the pathogenicity of S. aureus, as it may be present in sputum samples, in the absence of respiratory symptoms (Lyczak, et al, 2002;15:194) • However, bronchoalveolar samples from infants diagnosed with CF by newborn screening demonstrate that in approximately 30% S. aureus can be detected, and its presence is associated with a significant inflammatory response (Armstrong, et al, 1997;156:1197) • There remains controversy regarding the role of prophylactic anti-staph antibiotics • Infants diagnosed by newborn screening and treated with long-term flucloxacillin for the first two years of life had, less frequent cough, less S. aureus isolates, lower admission rates ( 5 v 19), for shorter periods (2.2 v 6.4 days), despite receiving half the number of antibiotic courses (in addition to the flocloxacillin) (Weaver et al, 1994;70:84)
S. aureus • A double blind placebo controlled trial of cephalexin as a long-term anti-staph prophylaxis started at mean age of 15 months, demonstrated reduced detection of S. aureus in airway cultures (6% v 30%), however there was no difference in clinical outcome measures, including lung function, x-ray scores, and nutrition. Most concerning was the increased detection of P. aeruginosa in the prophylactic cohort (26% v 13%) (Stutman, 2002;140:299) • Currently the UK CF trust recommends the use of flucloxacillin as anti-staph prophylaxis up to two years of age. When S. aureus is detected after this, a two week course of anti-staph antibiotics is commenced.
Pseudomonas Aeruginosa • P. aeruginosa is the most common chronic airway infection affecting patients with CF. The prevalence of infection increases with age. By the third decade up to 80% of patients are infected (Gibson et al, 2003;168:918) • Chronic P. aeruginosa infection is difficult to eradicate and is associated with more respiratory symptoms, worse general health (Pamukcu et al 1995;19:10), more rapid decline in lung function (Kerem et al, 1990;116:714), more rapid deterioration in CXR scores (Demko et al, 1995;48:1041) and significantly worse survival (Lyczak et al, 2002;15:194) (Emerson et al, 2002;34:91) • The ability of P. aeruginosa to change from a non-mucoid to mucoid phenotype by inducing bio-film formation, is an important feature in the establishment of chronic infection (Lyczak et al, 2002;15:194) • Bio-films protect the organism from destruction by both neutrophils and antibiotics (Drenkard et al, 2002;416:695) (Costerton et al, 1999;284:1318) • There are three different strategies for the antibacterial treatment of P. aeruginosa, depending on the clinical situation: first isolation, infective exacerbation, and chronic infection
1. First isolation • Initially P. aeruginosa isolates are non-mucoid, motile, and are present in low density, thus presenting an opportunity for eradication (Rosenfeld et al, 2001;32:356). • There is no consensus regarding the best eradication regime, but they all involve early aggressive treatment with anti-pseudomonal antibiotics • Three weeks of nebulised colistin (1MU bd if < 2 yo, and 2 MU bd if > 2 yo) with 2-3 weeks of oral ciprofloxacin is as effective (80%) in initial eradication as three months of nebulised colisitin with 3 weeks of oral ciprofloxacin, but the three month treatment regime increased the interval to re-infection from 9 to 18 months (Frederiksen et al 1997;23:330) (Taccetti et al, 2005;26:458)
1. First isolation • 28 days treatment with twice daily nebulised Tobramycin (300mg) for inhalation (TOBI) eradicated P. aeruginosa from 8 of 8 children less than 6 y/o, compared with just 1 of 13 children treated with placebo (Gibson et al, 2003;167:841) • Inhaled tobramycin (80mg) for one year eradicated P. aeruginosa from 14 of 15 patients (Ratjen et al, 2001;358:983) (Wiesemann et al, 1998;25:88) • When P. aeruginosa is detected again after eradication it is typically a new strain, rather than a re-emergence of partially treated original strain (Munck et al, 1998;32:288) (Taccetti et al, 2005;26:458)
2. Acute exacerbation • An acute exacerbation is characterised by an increase in respiratory symptoms and signs, along with a reduction in lung function - typically a drop in FEV1 > 10% (Dakin et al, 2001;31:436) (Marshall, 2004;169:781). Reduced energy, and appetite, along with weight loss often also present. • Minor exacerbations are typically treated with two weeks of oral ciprofloxacin (a quinolone), in addition to usual inhaled medications, and an increase in physiotherapy • More significant infections typically require an increase in physiotherapy, along with anti-pseudomonal IV antibiotics for 14 days • Usually two antibiotics are started; a beta-lactam antibiotic (e.g. ceftazidime, tazobactum + piperacillin, meropenum) plus an aminoglycoside (e.g. tobramycin, gentamicin, amikacin)
3. Chronic infection • Chronic P. aeruginosa infection is defined as the culture of P. aeruginosa in 2 or more airway samples over a six month period (UK CF Trust 2001) • Long-term use of nebulised antibiotics has been shown to be beneficial – it should be noted that this is long-term treatment, rather than prophylaxis • Overall, only approximately 10% of the nebulised drug is delivered to the lungs (Le Conte et al, 1993;147:1279) • Long-term nebulised tobramycin for inhalation (300mg twice daily) has been demonstrated to be safe, and to improve quality of life, increase lung function and reduce exacerbation rate (Ramsey et al, 1999;340:23) (Ramsey et al, 1993;328:1740) (Quittner et al, 2002;33:269) (Moss et al, 2002;121:55)
3. Chronic infection • Tobi is to be given with the Pari LC Plus nebuliser with Pari TurboBoy compressor • Nebulised Colistin has also been used for some time, particularly in Europe (Littlewood et al, 2000;94:632) • The effectiveness of long-term nebulised Colistin has been demonstrated, with improved symptoms, reduced bacterial load, and maintenance of lung function (Littlewood, et al, 1985;1:865) (Jensen et al, 1987;19:831) • However, there is some limited evidence that nebulised Tobramycin is superior to Colistin in maintaining lung function (Hodson et al, 2002;20:658)
Burkholderia Cepacia • Burkholderia is a genus which contains several species. Initially the species were phenotypically indistinguishable, but were identified through molecular genetic typing as distinct genomovars (I – IX) (Vandamme et al, 1997;47:1188) • The Burkholderia species can now be identified phenotypically, with the nine genomovars which cause lung disease in CF collectively referred to as the Burkholderia Cepacia Complex (Coenye et al, 2003;41:2797)
Burkholderia Cepacia Complex • B. cenocepacia (previously Genomovar III), and B. multivorans (previously Genomovar I) cause the majority of infections in the CF population • B. cenocepacia is particularly problematic • It is highly transmissible among CF patients • Approx 1/3 of patients infected will develop Cepacia Syndrome, which results in systemic signs of sepsis, weight loss, CXR infiltrates, a rapid decline in lung function, and early death within weeks, to months, despite maximal antimicrobial therapy. (Jones et al, 2004;59:948) • There are some patients who remain well despite the presence of B. cenocepacia in their airways, however the majority with chronic infection have a more rapid decline in lung function, with a worse prognosis, than those with chronic Pseudomonas infection (McCloskey et al, 2001;170:28) • It is typically resistant to most antibiotics, however, meropenum or ceftazidime, in combination with an aminoglycoside, may be useful, with co-trimoxazole added as a third agent (Aaron et al, 2000;161:1206)
Non-tuberculous mycobacterium • Mycobacterium avium complex (MAC) and Mycobacterium abscessus account for 70% and 15% respectively of non-tuberculous mycobacterial (NTM) infection in CF patients, with M. abscessus being the more virulent of the two (Olivier et al, 2003;167:828) • NTM tend to be transiently detected in the airways, with only a minority of patients developing clinically relevant disease. (Olivier et al, 2003;167:835) • The ATS guidelines for the diagnosis of NTM infection are: • Three sequential positive airway cultures • A decline in lung function • New changes on CT scanning (ATS, 1997;156:S1-25) • However, as NTM is often a co-infection with Pseudomonas, it is prudent to treat Pseudomonas infection first, and if there is no clinical improvement, to then start anti-NTM therapy • Rifampicin, ethambutol, azithromycin and ciprofloxacin have been shown to be useful. Imipenem, and amikacin (IV or nebulised) may also be used in difficult cases. Treatment should be continued for one year with negative sputum cultures. (Leitritz et al, 2004;139:209) (Forslow et al, 2003;92;910) IFN-gamma may also have a role, in some cases (Hallstrand et al, 2004;24:367).
ABPA • Aspergillus is a fungus which is commonly found in the airway samples of patients with CF (prevalence of 1 – 60%) (Stevens et al, 2003;37(suppl):S225) • On many occasions it is simply a non-pathological colonizer of the airway, however a hypersensitivity reaction may develop to its presence resulting in allergic bronchopulmonary aspergillosis (ABPA) • Diagnostic criteria for ABPA are : • The occurrence of asthma symptoms • New CXR changes • A four fold increase in IgE titres, or Ige > 500 iU/L • Increased specific IgE RAST to aspergillus or positive skin prick test • Eosinophilia > 500/mm3 • Positive sputum culture (CF UK Trust Antibiotic treatment for CF, 2nd Ed, 2002; available at www.cftrust.org.uk )
ABPA • Treatment • 0.5-1mg/kg/day Prednisolone for two weeks, with gradual tapering of the dose over 2 – 3 months • The role of anti-fungals is not clear, but treatment with itraconazole is often initiated with steroid treatment (Stevens et al, 2003;37(suppl):S225)
Stenotrophomonas maltophilia • Stenotrophomonas maltophilia does not appear to have a significant impact on lung function or mortality (Goss et al, 2002;166:356). • It does not appear to be transmitted between patients, and the majority of patients only have it transiently in their airways (Goss et al, 2004;59:955) • There are no clinical trials of antibiotic treatment for S. maltophilia, however as some patients may go on to develop chronic infection, first isolates should probably be treated. Equally patients who are symptomatic, and/or deteriorating, should have antibiotics prescribed which are effective against S. maltophilia. • S. maltophilia is resistant to a broad range of antibiotics. Ceftazidime may be useful, or IV piptazo and an aminoglycoside. Oral co-trimoxazole also has useful activity (Krueger et al, 2001;41:71).
Haemophilus Influenzae • Haemophilus influenzae is detected frequently in the airways of patients with CF, and is often detected early in the course of the disease (Armstrong et al 1997;156:1197) • Although there is little evidence that it causes significant pathology, it is however pathogenic in non-CF bronchiectasis (Bilton et al, 1995;8:948) • It is often associated with increased symptoms and when this is the case it should be treated with an oral antibiotic such as co-amoxiclav (Rayner et al, 1990;65:255)
Airway clearance • Daily physiotherapy forms the cornerstone of CF airway clearance • Historically postural drainage and percussion was used, although there are a wide number of modalities now available.
Percussion technique • Postural drainage involves placing the patient in positions that allows gravity to assist mucous clearance (there are 12 positions, although the head down position is now generally avoided because of concerns regard gastro-oesophageal reflux) • The technique requires an assistant to percus over the chest for 2-10 minutes, usually followed by deep breathing and huffing • The technique is now mostly used on infants and young children