Genome Variations & GWAS
320 likes | 712 Views
I519 Introduction to Bioinformatics, 2012. Genome Variations & GWAS. cause inherited diseases. Genome variations. underlie phenotypic differences. sequence variations can be used for gene mapping, definition of population structure, and performance of functional studies. 1000 Genomes Project.

Genome Variations & GWAS
E N D
Presentation Transcript
I519 Introduction to Bioinformatics, 2012 Genome Variations & GWAS
cause inherited diseases Genome variations underlie phenotypic differences sequence variations can be used for gene mapping, definition of population structure, and performance of functional studies.
1000 Genomes Project • An international collaboration to produce an extensive public catalog of human genetic variation, including SNPs and structural variants, and their haplotype contexts. This resource will support genome-wide association studies and other medical research studies. • The genomes of about 2500 unidentified people from about 25 populations around the world will be sequenced using next-generation sequencing technologies. • Results of the pilot phase of the project published in Nature Volume: 467, Pages: 1061–1073, 2010
look at multiple sequences from the same genome region • use base quality values to decide if mismatches are true polymorphisms or sequencing errors How do we find sequence variations?
Automated polymorphism discovery PolyBayes (Ref: A general approach to single-nucleotide polymorphism discovery, Marth et al., Nature Genetics, 1999) Determine if a genetic difference is due to sequencing error or it is a real SNP by using base quality values in a rigorous, Bayesian scheme to compare sequences of arbitrary quality standards. PYROBAYES (Ref: An improved base-caller for SNP discovery in pyrosequences. Nature Methods. 2008;5:179-81.)
SNP functional categories • coding nonsynonymous • Missense, nonsense, frame shift • coding synonymous • Intronic • splice site • mRNA utr • 5' utr or 3' utr • (gene) locus region (5’ or 3’ to the gene) • ‘near gene’ usually means within ~2000bp of gene • genomic/extragenic (distant from any gene)
SNP nomenclature • The Human Genome Variation Society (http://www.hgvs.org/mutnomen/recs.html) has proposed some guidelines for SNP nomenclature, but at the moment, there is minimal consistency. • Different sources will refer to the same SNP in different ways • While dbSNP identifiers (rs#12345678) are becoming common, they are not required of publishing authors and not used in all cases.
SNPs at base-pair level • The base-pair change is given in various forms: A/C T→G C>T 432G>C T73C The HGVS nomenclature recommendations: "c." for a coding DNA sequence (like c.76A>T) "g." for a genomic sequence (like g.476A>T) "m." for a mitochondrial sequence (like m.8993T>C "r." for an RNA sequence (like r.76a>u)
dbSNP • SNP database from NCBI, build 130 contains 63,751,769 refSNP clusters (19,576,037 validated) • dbSNP contains: • Single nucleotide substitutions • Small insertion/deletion polymorphism • Microsatellite repeats
dbSNP content The SNP database has two major classes of content: • Submitted data, i.e., original observations of sequence variation: Submitted SNPs (SS) with ss# (e.g, ss5586300) • Reference Cluster ID: Computed/curated data (Ref SNP with rs#, e.g., rs25) • Ref SNP • Ref SNP Clusters define a non-redundant set of SNPs • Ref SNP clusters may contain multiple submitted SNPs
Reference SNP clusters • Ref SNP clusters are computer-generated and curated by NCBI staff • Ref SNP Clusters define a non-redundant set of SNPs • All individual SNPs submitted by a researcher are given a submitter SNP number (ss#) and then redundant (repetitive) submitter SNPs are combined into a RefSNP cluster record, with a unique rs# • Ref SNP clusters may contain multiple submitted SNPs
Promises of SNPs • Each person's SNP pattern is unique • Most SNPs are not responsible for a disease state. But they can be located near a gene associated with a certain disease. So SNPs may serve as biological markers for pinpointing a disease on the human genome map. • Application of association study can detect differences between the SNP patterns of two groups (control-disease), thereby indicating which pattern is most likely associated with the disease-causing gene. • Using SNPs to study the genetics of drug response will help in the creation of "personalized" medicine.
Annotation of SNPs • A straightforward and reliable method based on physical and comparative considerations that estimates the impact of an amino acid replacement on the three-dimensional structure and function of the protein (~20% of common human non-synonymous SNPs predicted to be deleterious). Ref: Human Molecular Genetics, 2001, Vol. 10, No. 6 591-597 • SIFT: predicting amino acid changes that affect protein function (used to distinguish between functionally neutral and deleterious amino acid changes in mutagenesis studies and on human polymorphisms). Ref: Nucleic Acids Research, 2003, Vol. 31, No. 13 3812-3814 • Review: Next generation tools for the annotation of human SNPs by Rachel Karchin, Briefings in Bioinformatics 2009 10(1):35-52
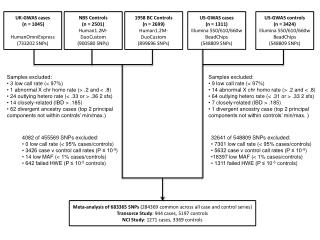
Genome-wide association study (GWAS) • A genome-wide association study is an approach that involves rapidly scanning markers across the complete sets of DNA, or genomes, of many people to find genetic variations associated with a particular disease. (http://www.genome.gov/20019523) • If genetic variations are more frequent in people with the disease, the variations are said to be "associated" with the disease. • Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls (Nature 447, 661-678) • Validating, augmenting and refining genome-wide association signals (Nature Reviews Genetics 10, 318-329, 2009)
The underlying rationale for GWAS • 'common disease, common variant' hypothesis, positing that common diseases are attributable in part to allelic variants present in more than 1–5% of the population • SNP genotyping chips – common variants • most common variants individually or in combination confer relatively small increments in risk (1.1–1.5-fold) and explain only a small proportion of heritability. E.g. at least 40 loci have been associated with human height (with an estimated heritability of about 80%), yet they explain only about 5% of phenotypic variance Ref: Finding the missing heritability of complex diseases; Nature461, 747-753, 2009
Explaining missing heritability • Rare variants; • variants of low minor allele frequency (MAF; 0.5% < MAF < 5%), or of rare variants (MAF < 0.5%) • Rare variants are not sufficiently frequent to be captured by current GWA genotyping arrays • And they don’t carry sufficiently large effect sizes to be detected by classical linkage analysis in family studies • The primary technology for the detection of rare SNPs is sequencing, which may target regions of interest, or may examine the whole genome. • Structural variation, including copy number variants (CNVs, such as insertions and deletions) and copy neutral variation (such as inversions and translocations)
Feasibility of identifying genetic variants by risk allele frequency and strength of genetic effect (odds ratio). TA Manolio et al. Nature461, 747-753 (2009) doi:10.1038/nature08494
Genome-wide significance • Associations that have been identified from a single GWA data set rarely have definitive statistical support. p values of <10-7 are required for genome-wide significance. A p value of approximately 10-7 in the GWA setting corresponds to a p value of approximately 0.05 for a traditional, classical epidemiological study in which only one hypothesis is being tested. • Statistical significance for genomewide studies • PNAS 100(16):9440-9445, 2003 • q value; similar to the well known p value, except it is a measure of significance in terms of the false discovery rate rather than the false positive rate.
Analysis methods (a) is a baseline analysis; (b)-(e) apply further prior hypotheses
Chi-square statistic tests Observed Expected O1=a, E1=nAAncase/n, and so on
Population stratification • Population stratification is the presence of a systematic difference in allele frequencies between subpopulations in a population possibly due to different ancestry. • Case control association studies assume that any difference in the SNP genotypes between the cases and controls is due solely to their difference in disease status, but not difference in their genetic background. • Potential population stratification needs to be corrected in association studies
GWAS vs genetic linkage method • Genetic linkage combined with positional cloning leads to the finding of gene mutations that are involved with monogenic disease, such as cystic fibrosis and Huntington's disease. These mutations most likely alter the amino acid sequence of protein. • Most loci that have been discovered through genome-wide association analysis do not map to amino acid changes in proteins (with a few important exceptions). They are predicted to affect gene expression.
Readings • Bioinformatics challenges for genome-wide association studies • Bioinformatics (2010) 26 (4): 445-455 • Finding the missing heritability of complex diseases • Nature (2009) 461, 747-753