Download
1 / 1
10 likes | 124 Views
This study presents innovative quantum mechanical approaches utilizing Lanczos and Chebyshev iterations to calculate unimolecular dissociation rates across the full thermal range of angular momenta. The challenges of high angular momentum calculations (J > 0) are addressed using parallel computing strategies, enabling effective computation of resonance states in the HO2 system, up to J = 50. Our findings demonstrate significant agreements between quantum mechanical rates and statistical adiabatic channel method results, highlighting the efficiency of our methodologies in complex unimolecular reaction dynamics.

E N D
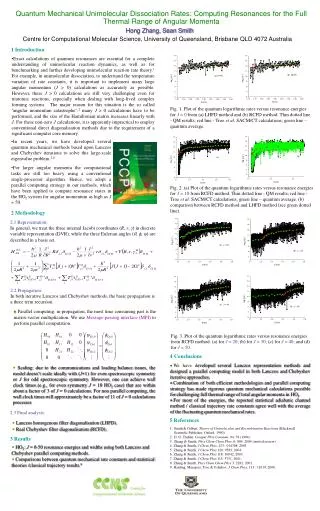
Quantum Mechanical Unimolecular Dissociation Rates: Computing Resonances for the Full Thermal Range of Angular Momenta Hong Zhang, Sean Smith Centre for Computational Molecular Science, University of Queensland, Brisbane QLD 4072 Australia 1 Introduction • Exact calculations of quantum resonances are essential for a complete understanding of unimolecular reaction dynamics, as well as for benchmarking and further developing unimolecular reaction rate theory.1 For example, in unimolecular dissociation, to understand the temperature variation of rate constants, it is important to implement many large angular momentum (J > 0) calculations as accurately as possible. However, these J > 0 calculations are still very challenging even for triatomic reactions, especially when dealing with long-lived complex forming systems. The major reason for this situation is the so called ‘angular momentum catastrophe’:2 many J > 0 calculations have to be performed, and the size of the Hamiltonian matrix increases linearly with J. For these non-zero J calculations, it is apparently impractical to employ conventional direct diagonalisation methods due to the requirement of a significant computer core memory. Fig. 1. Plot of the quantum logarithmic rates versus resonance energies for J = 0 from (a) LHFD method and (b) RCFD method. Thin dotted line - QM results; red line - Troe et al. SACM/CT calculations; green line – quantum average. • In recent years, we have developed several quantum mechanical methods based upon Lanczos and Chebyshev iterations to solve this large-scale eigenvalue problem.3-8 • For larger angular momenta the computational tasks are still too heavy using a conventional single-processor algorithm. Hence, we adopt a parallel computing strategy in our methods, which have been applied to compute resonance states in the HO2 system for angular momentum as high as J = 50. Fig. 2. (a) Plot of the quantum logarithmic rates versus resonance energies for J = 10 from RCFD method. Thin dotted line - QM results; red line - Troe et al. SACM/CT calculations; green line – quantum average. (b) comparison between RCFD method and LHFD method (see green dotted line). 2 Methodology 2.1 Representation: In general, we treat the three internal Jacobi coordinates (R, r, ) in discrete variable representation (DVR), while the three Eulerian angles (, , ) are described in a basis set. 2.2 Propagation: In both iterative Lanczos and Chebyshev methods, the basic propagation is a three term recursion. Parallel computing: in propagation, the most time consuming part is the matrix-vector multiplication. We use Message-passing interface (MPI) to perform parallel computation. Fig. 3. Plot of the quantum logarithmic rates versus resonance energies from RCFD method. (a) for J = 20; (b) for J = 30;(c) for J = 40; and (d) for J = 50. 4 Conclusions • We have developed several Lanczos representation methods and designed a parallel computing model in both Lanczos and Chebyshev iterative approaches. • Combination of both efficient methodologies and parallel computing strategy has made rigorous quantum mechanical calculations possible for challenging full thermal range of total angular momenta in HO2. • For most of the energies, the reported statistical adiabatic channel method / classical trajectory rate constants agree well with the average of the fluctuating quantum mechanical rates. Scaling: due to the communications and loading balance issues, the model doesn’t scale ideally with (J+1) for even spectroscopic symmetry or J for odd spectroscopic symmetry.However, one can achieve wall clock times (e.g., for even symmetry J = 10 HO2 case) that are within about a factor of 3 of J = 0 calculations. For non parallel computing, the wall clock times will approximately be a factor of 11 of J = 0 calculationsprocesses 2.3 Final analysis: 5 References Lanczos homogenous filter diagonalisation (LHFD). Real Chebyshev filter diagonalisation (RCFD). • Smith & Gilbert, Theory of Unimolecular and Recombination Reactions (Blackwell Scientific Publisher, Oxford, 1990). • D. G. Truhlar, Comput Phys Commun, 84: 78 (1994). • Zhang & Smith, Phys Chem Chem Phys, 6: 884, 2004 (invited review). • 4. Zhang & Smith, J Chem Phys, 123: 014308, 2005. • 5. Zhang & Smith, J Chem Phys 120: 9583, 2004. • 6. Zhang & Smith, J Chem Phys 118: 10042, 2003. • 7. Zhang & Smith, J Chem Phys 115: 5751, 2001. • 8. Zhang & Smith, Phys Chem Chem Phys 3: 2282, 2001. • 9. Harding, Maergoiz, Troe & Ushakov, J Chem.Phys, 113 : 11019, 2000. 3 Results HO2: J = 0-50 resonance energies and widths using both Lanczos and Chebyshev parallel computingmethods.Comparisons between quantum mechanical rate constants and statistical theories /classical trajectory results.9



















